Bệnh tan máu bẩm sinh Thalassemia
Bệnh tan máu bẩm sinh hay thiếu máu tán huyết Thalassemia là một nhóm bệnh di truyền về máu khiến cơ thể không thể sản xuất đủ lượng hemoglobin cần thiết gây nên tình trạng thiếu máu tán huyết. Hemoglobin nằm trong các tế bào hồng cầu, có chức năng mang oxy từ phổi đến khắp cơ thể. Không có đủ hemoglobin dẫn đến thiếu máu, khiến người bệnh cảm thấy mệt mỏi, yếu ớt và khó thở.
Có 2 dạng bệnh chính gồm:
Đôi khi, beta thalassemia có thể xảy ra cùng với một đột biến gen khác gây bệnh hồng cầu hình liềm (SCD), gọi là bệnh beta thalassemia hình liềm hoặc beta thalassemia S.
Biểu hiện lâm sàng
Các triệu chứng bệnh thalassemia phụ thuộc vào thể alpha hay beta thalassemia và mức độ nghiêm trọng của bệnh. Các triệu chứng có thể từ nhẹ đến nặng, một số người có ít hoặc không có triệu chứng. Các triệu chứng của bệnh thalassemia có thể bao gồm một hoặc nhiều triệu chứng sau:
- Xanh xao
- Mệt mỏi, ít năng lượng
- Lâng lâng hoặc khó thở
- Chán ăn
- Nước tiểu đậm
- Vàng da (vàng da và lòng trắng của mắt)
- Trẻ chậm lớn và dậy thì muộn
- Dị dạng xương mặt
- Phình bụng
Alpha thalassemia có thể gây hại cho phụ nữ mang thai, làm thai chết lưu.
Trẻ em sinh ra bị bệnh thalassemia có thể xuất hiện các dấu hiệu của bệnh ngay lập tức, hoặc các triệu chứng có thể xuất hiện muộn hơn. Hầu hết các dấu hiệu và triệu chứng thường xuất hiện trong vòng 2 năm đầu đời. Nếu con bị chậm phát triển, cần kiểm tra xem con có mắc bệnh thalassemia hay không. Bệnh thalassemia không được điều trị có thể gây suy tim và nhiễm trùng.
Nhiều người mắc bệnh thalassemia có cuộc sống khỏe mạnh. Nhưng nó có thể gây ra tình trạng sức khỏe nghiêm trọng cần được điều trị. Thalassemia thể nặng có thể tử vong.
Các biến chứng
Bệnh thalassemia thể nặng có thể dẫn đến các biến chứng sức khỏe nghiêm trọng bao gồm:
- Các vấn đề về xương. Thalassemia có thể làm cho tủy xương mở rộng, khiến xương trở nên dễ vỡ và gãy.
- Vấn đề về tim. Các dạng thalassemia nghiêm trọng có thể gây ra các vấn đề về tim, bao gồm cả suy tim.
- Lá lách to. Lá lách là một cơ quan nằm phía trên dạ dày và dưới xương sườn ở bên trái, giúp cơ thể chống lại nhiễm trùng và loại bỏ các tế bào già cỗi hư hỏng. Ở những người mắc bệnh thalassemia, các tế bào máu chết nhanh hơn, khiến lách hoạt động nhiều hơn. Kết quả là lách to ra và có thể làm cho tình trạng thiếu máu trầm trọng hơn. Lá lách có thể cần phải được cắt bỏ nếu phát triển quá lớn.
- Nhiễm trùng. Những người mắc bệnh thalassemia có khả năng bị nhiễm trùng cao hơn những người khác, đặc biệt sau khi cắt lá lách.
- Tăng trưởng chậm. Trẻ thiếu máu có thể chậm lớn và dậy thì muộn.
- Quá nhiều sắt trong máu. Có thể gây ra các vấn đề về tim, gan và các cơ quan khác trong cơ thể.
Độ phổ biến
Khoảng 100.000 trẻ sơ sinh trên toàn thế giới được sinh ra mắc bệnh thalassemia mức nặng, cả bé trai và bé gái. Bệnh phổ biến nhất ở các nước như Ý, Hy Lạp, các nước khu vực Trung Đông, Châu Á hoặc Châu Phi.
Theo số liệu năm 2019 của Bệnh viện Huyết học và truyền máu Trung Ương, tại Việt Nam ước tính khoảng 13 triệu người mang gen bệnh thể ẩn, hơn 2000 trẻ sinh ra hàng năm bị bệnh mức nặng phải truyền máu và thải sắt suốt đời. Do đó, hiện nay tất cả trẻ sơ sinh đều được khuyến cáo lấy máu gót chân làm sàng lọc bệnh tan máu Thalassemia trong vòng 48 giờ sau khi sinh.
Nguyên nhân
Trong cơ thể có ba loại tế bào máu: hồng cầu, bạch cầu và tiểu cầu. Các tế bào hồng cầu chứa hemoglobin, một loại protein chứa sắt có chức năng vận chuyển oxy từ phổi đến tất cả các bộ phận của cơ thể.
Hemoglobin có hai loại chuỗi protein: alpha globin và beta globin. Nếu cơ thể không tạo đủ các chuỗi protein này hoặc chúng bất thường, các tế bào hồng cầu sẽ không hình thành hoặc không mang đủ oxy. Cơ thể sẽ không hoạt động tốt nếu các tế bào hồng cầu không tạo đủ hemoglobin khỏe mạnh.
Một người bị mắc bệnh thalassemia khi các gen kiểm soát tạo ra protein hemoglobin bị mất hoặc đột biến.
Thể bệnh thalassemia mắc phải phụ thuộc vào loại gen đột biến thừa hưởng từ cha mẹ và số lượng gen thừa hưởng.
- Alpha-thalassemia: Gen HBA1 và HBA2 mang các hướng dẫn tạo ra alpha globin (một trong hai loại protein tạo nên hemoglobin). Mất một số hoặc tất cả các gen HBA1 và HBA2 gây thiếu alpha globin, dẫn đến bệnh alpha-thalassemia.
- Beta-thalassemia: Gen HBB mang các hướng dẫn để tạo ra beta globin (một trong hai loại protein tạo nên hemoglobin). Những thay đổi (đột biến) trong gen HBB dẫn đến giảm nồng độ beta globin và gây ra bệnh beta thalassemia.
Thalassemia có tính di truyền, nghĩa là nó được truyền từ cha mẹ sang con cái thông qua gen. Nếu trong gia đình có người mắc bệnh thalassemia, có thể nói người đó có tiền sử gia đình mắc bệnh thalassemia, các thành viên khác nên làm xét nghiệm sàng lọc gen để biết chính xác tình trạng.
Chẩn đoán
Bệnh thalassemia mức trung bình và nặng thường được chẩn đoán sau khi chào đời. Trẻ sinh ra có các dấu hiệu và triệu chứng đặc trưng bao gồm thiếu máu trầm trọng thường xảy ra trong vòng 2 năm đầu đời. Những người mắc bệnh thalassemia dạng nhẹ hơn thường không có biểu hiện rõ ràng, có thể được sàng lọc bằng xét nghiệm máu thông thường, nếu kết quả ra nguy cơ cao, cần được chẩn đoán cuối cùng bằng xét nghiệm gen Thalassemia.
Xét nghiệm sàng lọc
- Công thức máu toàn phần đo lượng hemoglobin và các loại tế bào máu khác nhau trong một mẫu máu. Người bệnh thalassemia thường có ít tế bào hồng cầu khỏe mạnh và ít hemoglobin hơn những người bình thường. Những người mắc alpha thalassemia hoặc beta thalassemia ở thể nhẹ có thể có hồng cầu nhỏ hơn bình thường.
- Điện di huyết sắc tố (hemoglobin) đo các loại hemoglobin trong máu. Những người mắc bệnh thalassemia có vấn đề với chuỗi protein alpha hoặc beta globin của hemoglobin.
- Xét nghiệm lượng sắt trong máu để xác định tình trạng thiếu máu do thiếu sắt hay bệnh thalassemia. Thiếu máu do thiếu sắt xảy ra nếu cơ thể không có đủ sắt để tạo ra hemoglobin. Thiếu máu trong bệnh thalassemia xảy ra do vấn đề với chuỗi alpha globin hoặc beta globin của hemoglobin, không phải do thiếu sắt.
Xét nghiệm chẩn đoán
Bởi vì bệnh thalassemia có nguyên nhân do đột biến gen, xét nghiệm gen Thalassemia giúp xác định chính xác có tồn tại gen đột biến hay không, nếu có thì đó là loại đột biến gì. Dựa vào kết quả này, bác sĩ có thể kết luận chính xác tình trạng bệnh và đưa ra các liệu pháp điều trị nếu cần thiết.
Thalassemia có thể chuẩn đoán cho thai nhi. Nếu cặp vợ chồng cùng mang đột biến gây bệnh thalassemia, có thể kiểm tra bằng các xét nghiệm trước sinh như:
- Sinh thiết gai nhau (CVS): kiểm tra mô từ nhau thai để xem liệu trẻ có bị bệnh di truyền hay không. Xét nghiệm được thực hiện khi thai được 10 đến 13 tuần tuổi.
- Chọc ối: Xét nghiệm này lấy một ít nước ối từ bụng mẹ để kiểm tra các dị tật bẩm sinh và bệnh di truyền. Xét nghiệm được thực hiện khi thai được 16 đến 22 tuần tuổi.
Điều trị
Liệu pháp điều trị thalassemia tùy thuộc vào loại bệnh và mức độ nghiêm trọng của bệnh. Những người mang gen bệnh hoặc bị thalassemia mức nhẹ có triệu chứng không rõ ràng hoặc đôi khi không có triệu chứng, nên có thể cần ít hoặc không cần điều trị
Hiện nay, có ba phương pháp điều trị cho các dạng thalassemia vừa và nặng, bao gồm: truyền máu, thải sắt và bổ sung axit folic. Các phương pháp điều trị khác đã được phát triển hoặc đang được thử nghiệm, tuy nhiên ít được sử dụng hơn.
Phương pháp thông dụng
Truyền máu
Truyền máu là phương pháp điều trị chính cho những người mắc bệnh thalassemia và hoặc nặng. Phương pháp điều trị này cung cấp hemoglobin bình thường cho người bệnh.
Các tế bào hồng cầu chỉ sống trong khoảng 120 ngày. Vì vậy, người bệnh cần truyền máu nhiều lần để đảm bảo đủ lượng tế bào hồng cầu khỏe mạnh.
Nếu chỉ bị bệnh hemoglobin H hoặc bệnh beta thalassemia thể trung gian, người bệnh có thể cần truyền máu trong một vài trường hợp như bị nhiễm trùng, bị bệnh hoặc khi tình trạng thiếu máu nghiêm trọng đến mức gây mệt mỏi.
Nếu mắc bệnh beta thalassemia thể nặng (thiếu máu Cooley), người bệnh sẽ cần truyền máu thường xuyên (thường là 2 đến 4 tuần một lần). Việc truyền máu này sẽ giúp duy trì lượng hemoglobin và hồng cầu bình thường.
Truyền máu cho phép người bệnh hoạt động bình thường và sống đến tuổi trưởng thành. Phương pháp điều trị này là cứu cánh cho nhiều người bệnh, tuy nhiên tốn kém và có nguy cơ nhiễm trùng và virus (ví dụ: viêm gan).
Thải sắt
Hemoglobin trong hồng cầu là một loại protein giàu chất sắt. Do đó, truyền máu thường xuyên có thể dẫn đến tích tụ sắt trong máu, gọi là dư thừa sắt. sắt tích tụ nhiều làm tổn thương gan, tim và các bộ phận khác của cơ thể.
Để ngăn ngừa tình trạng này, các bác sĩ sử dụng liệu pháp thải sắt để loại bỏ lượng sắt dư thừa ra khỏi cơ thể. Hai loại thuốc được sử dụng cho liệu pháp thải sắt.
- Deferoxamine: một loại thuốc dạng lỏng được truyền qua đường tĩnh mạch vào cơ thể. Liệu pháp này cần thời gian và có thể gây đau nhẹ. Các tác dụng phụ bao gồm các vấn đề về thị lực và thính giác.
- Deferasirox: là thuốc dạng viên uống mỗi ngày một lần. Các tác dụng phụ bao gồm nhức đầu, buồn nôn (cảm giác đau bụng), nôn mửa, tiêu chảy, đau khớp và mệt mỏi.
Bổ sung axit folic
Axit folic là một loại vitamin B giúp các tế bào hồng cầu khỏe mạnh. Bác sĩ có thể đề nghị bổ sung axit folic ngoài việc điều trị bằng truyền máu và / hoặc liệu pháp thải sắt.
Phương pháp khác
Một số phương pháp điều trị bệnh thalassemia khác đã được phát triển hoặc đang được thử nghiệm, nhưng chúng ít được sử dụng hơn.
Ghép tế bào gốc
Tế bào gốc máu là các tế bào bên trong tủy xương, có chức năng tạo ra các tế bào hồng cầu và các loại tế bào máu khác. Ghép tế bào gốc máu (từ một người hiến tặng) thay thế các tế bào gốc bị lỗi giúp cơ thể người bệnh tự tạo ra các tế bào hồng cầu khỏe mạnh.
Cấy ghép tế bào gốc là phương pháp điều trị duy nhất có thể chữa khỏi bệnh thalassemia. Tuy nhiên, rất khó tìm được người hiến tặng phù hợp và quy trình thực hiện khá mạo hiểm.
Dạng di truyền
Thalassemia di truyền theo kiểu lặn trên nhiễm sắc thể thường, tuy nhiên cơ chế khá phức tạp vì nhiều gen liên quan đến quá trình sản xuất hemoglobin.
Cơ chế di truyền của alpha thalassemia rất phức tạp do đột biến ở 2 gen khác nhau (HBA1 và HBA2). Mỗi người có 2 bản sao của gen HBA1 và 2 bản sao của gen HBA2 trong mỗi tế bào. Đối với mỗi gen, một bản sao được thừa hưởng từ mẹ và một bản sao được thừa hưởng từ cha. Nếu cha hoặc mẹ có ít nhất một bản sao bị đột biến gây bệnh, con sẽ có nguy cơ mắc alpha thalassemia. Tuy nhiên, mức độ nghiêm trọng của từng trường hợp phụ thuộc vào số lượng bản sao gen bị đột biến.
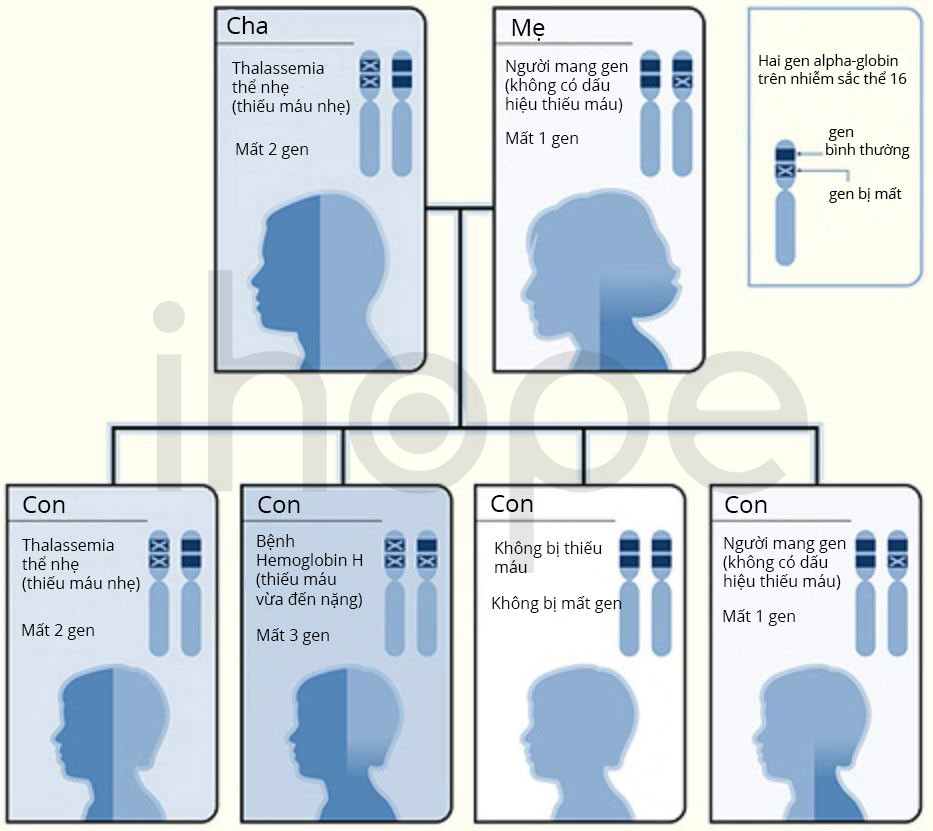
Nguồn: NHLBI
Hầu hết người bệnh beta thalassemia đều có đột biến ở cả hai bản sao của gen HBB trong mỗi tế bào. Cha mẹ của người bệnh mang một bản sao đột biến của gen nên được gọi là người mang gen bệnh thể ẩn hay mang gen lặn. Người mang gen lặn thường không có dấu hiệu hoặc triệu chứng của bệnh, một số có biểu hiện thiếu máu nhẹ. Khi hai người mang gen bệnh kết hôn, mỗi người con có 25% nguy cơ mắc bệnh nặng, 50% nguy cơ mang gen lặn như bố và mẹ và 25% khỏe mạnh hoàn toàn.
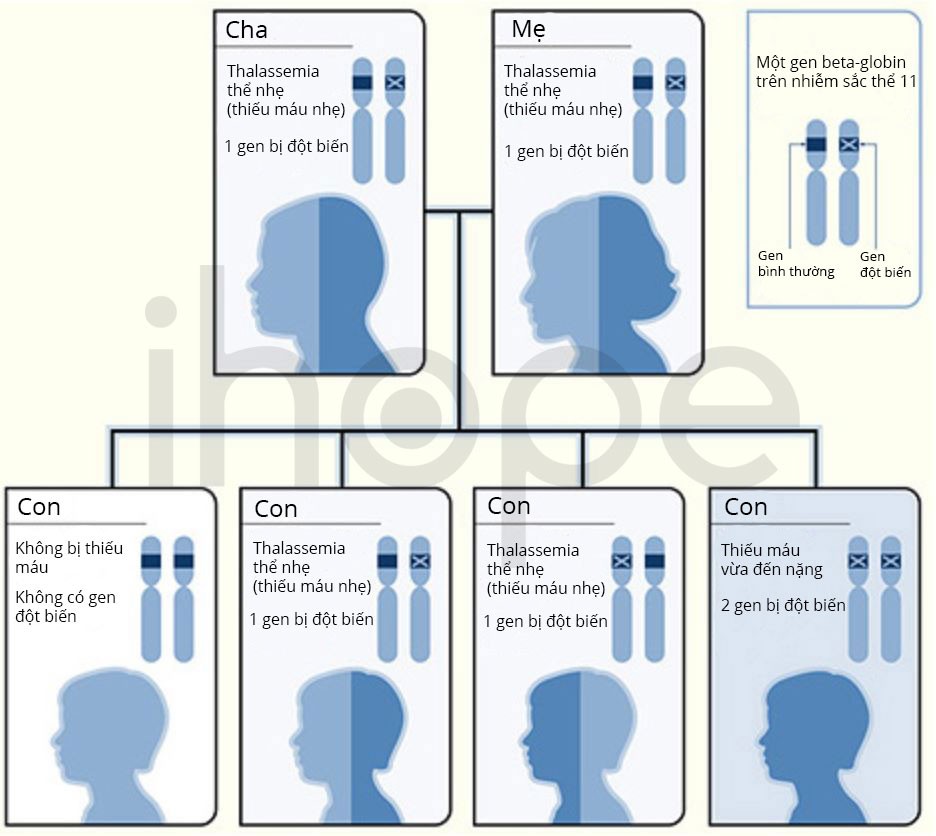
Nguồn: NHLBI
Phòng ngừa
Bệnh tan máu Thalassemia khó điều trị, nhưng có thể chủ động phòng ngừa bằng xét nghiệm sàng lọc gen lặn cho các cặp vợ chồng trước khi kết hôn hay sinh con. Nếu chỉ vợ hoặc chồng mang đột biến gen bệnh (thể ẩn) thì có thể sinh con. Trường hợp cả vợ chồng cùng mang đột biến gen bệnh giống nhau, cần chọc ối tại tuần 16-22 để xét nghiệm chẩn đoán thai nhi.
Các tên gọi khác
Các loại bệnh thalassemia có tên cụ thể liên quan đến mức độ nghiêm trọng của rối loạn.
Alpha Thalassemia
- Người mang gen bệnh alpha thalassemia
- Alpha thalassemia thể nhẹ
- Bệnh hemoglobin H
- Alpha thalassemia thể nặng (còn gọi là phù thai)
Beta Thalassemia
- Beta thalassemia thể nhẹ
- Beta thalassemia thể trung gian
- Bệnh beta thalassemia thể nặng (còn gọi là bệnh thiếu máu Cooley hoặc bệnh thalassemia beta-0 (ß0))
- Beta-plus (ß +) thalassemia
References
- U.S National Library of Medicine. Thalassemia. Retrieved April 1, 2021 from https://medlineplus.gov/thalassemia.html
- National Human Genome Research Institute. About Thalassemia. Retrieved April 1, 2021 from https://www.genome.gov/Genetic-Disorders/Thalassemia
- Centers for Disease control and Prevention. Thalassemia. Retrieved April 1, 2021 from https://www.cdc.gov/ncbddd/thalassemia/facts.html
- American Aademy of Family Physicians. Thalassemia. Retrieved april 1, 2021 from https://familydoctor.org/condition/thalassemia/
- March of Dimes. Thalassemia. Retrieved april 1, 2021 from https://www.marchofdimes.org/complications/thalassemia.aspx
- National Heart, Lung and Blood Institute. Thalassemia. Retrieved april 1, 2021 from https://www.nhlbi.nih.gov/health-topics/thalassemias