Hemoglobin là thành phần quan trọng đối với cơ thể con người. Chúng đảm nhận vị trí chủ chốt trong quá trình vận chuyển oxy từ phổi đến tế bào và mô, từ đó cơ thể được cung cấp năng lượng cần thiết để duy trì sự sống và hoạt động. Hemoglobin kết hợp với oxy trong phổi, sau đó chúng theo dòng máu vận chuyển oxy từ phổi đến những vị trí khác trong cơ thể.
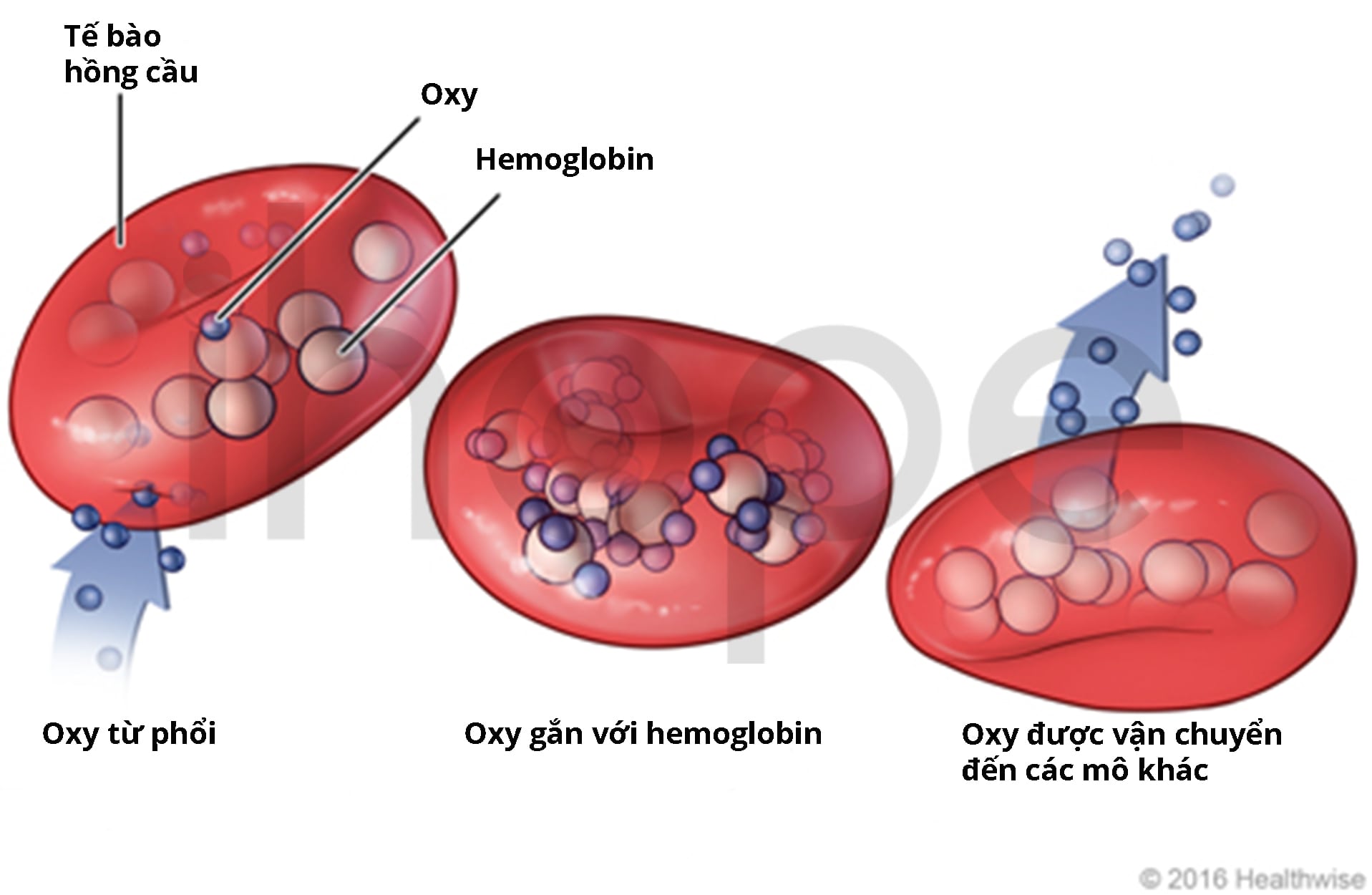
Nguồn: Healthwise
Ngoài ra, hemoglobin còn thực hiện nhiều chức năng quan trọng khác như:
- Vận chuyển CO2: hemoglobin tham gia vận chuyển carbon dioxide (CO2) ra khỏi tế bào và mô.
- Duy trì độ cân bằng pH: hemoglobin giúp điều chỉnh và duy trì pH của máu.
- Duy trì màu đỏ của máu: hemoglobin chứa sắt—yếu tố tạo nên màu đỏ đặc trưng của máu. Do đó, hemoglobin có thể giúp phát hiện nhiều bệnh lý về máu, vd thiếu máu nhược sắt.
- Kiểm soát quá trình oxy hóa: hemoglobin có khả năng chuyển đổi giữa trạng thái oxy hóa (oxyhemoglobin) và trạng thái giảm oxy hóa (deoxyhemoglobin) tùy thuộc vào nồng độ oxy trong môi trường xung quanh. Đặc điểm này giúp kiểm soát lưu lượng máu và cung cấp oxy đến bộ phận nào thiếu oxy trong cơ thể.
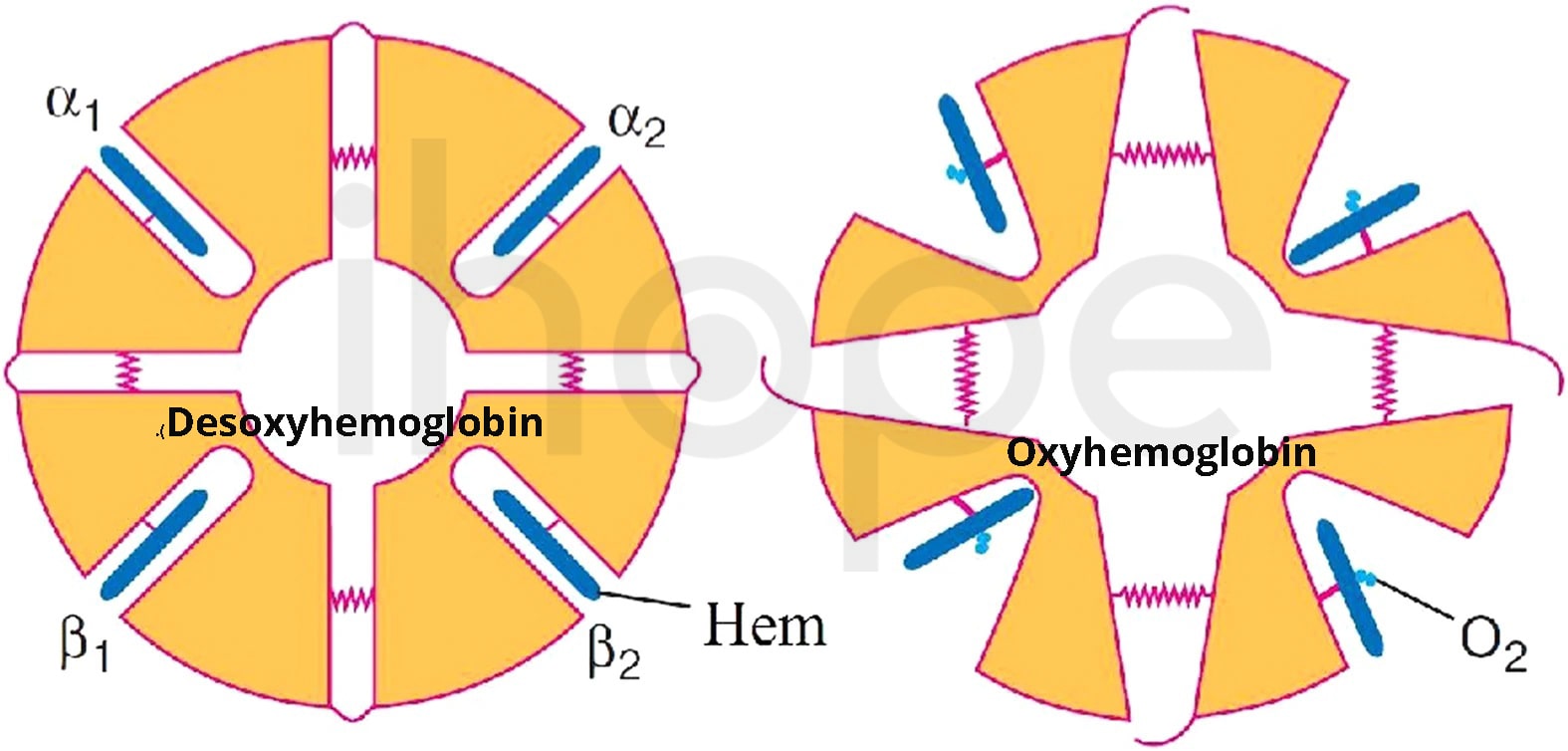
Nguồn: Scientific Reports
Do tầm quan trọng của hemoglobin, bất kỳ thay đổi hay rối loạn nào liên quan đến chúng đều có thể gây ra nhiều vấn đề sức khỏe từ nhẹ đến nặng.
Cấu trúc của hemoglobin
Hemoglobin là protein quan trọng có chức năng vận chuyển O2 và CO2 giữa các mô và tế bào trong cơ thể. Cấu trúc hemoglobin có hai phần bao gồm đơn vị globin và nhân heme gắn với globin.
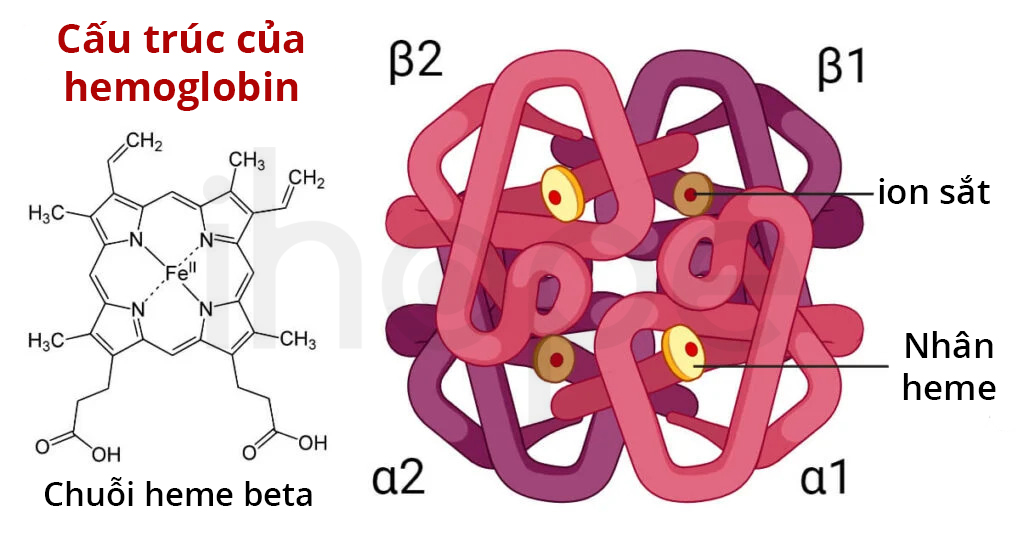
Nguồn: Microbe Notes
Đơn vị globin
Hai loại đơn vị globin chính có ký hiệu là α-globin và β-globin. Mỗi loại globin chứa một chuỗi polypeptide có số lượng và trình tự axit amin đặc trưng. Polypeptide là chuỗi bao gồm nhiều axit amin liên kết với nhau thông qua liên kết peptide. Bốn globin dạng monomere kết hợp với nhau tạo thành một phân tử hemoglobin dạng tetramere.
Tuỳ thuộc vào sự sắp xếp giữa các chuỗi globin sẽ tạo thành những loại hemoglobin khác nhau. Thông thường globin kết hợp với nhau theo nguyên tắc giống nhau từng đôi một và xếp đối xứng. Mỗi đơn vị globin trong hemoglobin có khả năng kết hợp với một phân tử oxy. Do đó, một hemoglobin có khả năng vận chuyển tối đa bốn phân tử oxy.
Nhân heme
Cấu trúc của nhân heme bao gồm một vòng porphyrin liên kết với ion sắt (Fe2+), ion sắt sẽ gắn với chuỗi globin. Khi oxy hiện diện gần đó, chúng sẽ kết hợp với nhân heme trong một đơn vị globin, từ đó hình thành oxyhemoglobin màu đỏ sáng và có khả năng vận chuyển oxy. Khi oxy giải phóng ra khỏi heme, hemoglobin trở lại thành dạng deoxyhemoglobin. Đây là cơ chế quan trọng đối với quá trình hô hấp để cung cấp oxy cho nhiều cơ quan trong cơ thể và loại bỏ CO2.
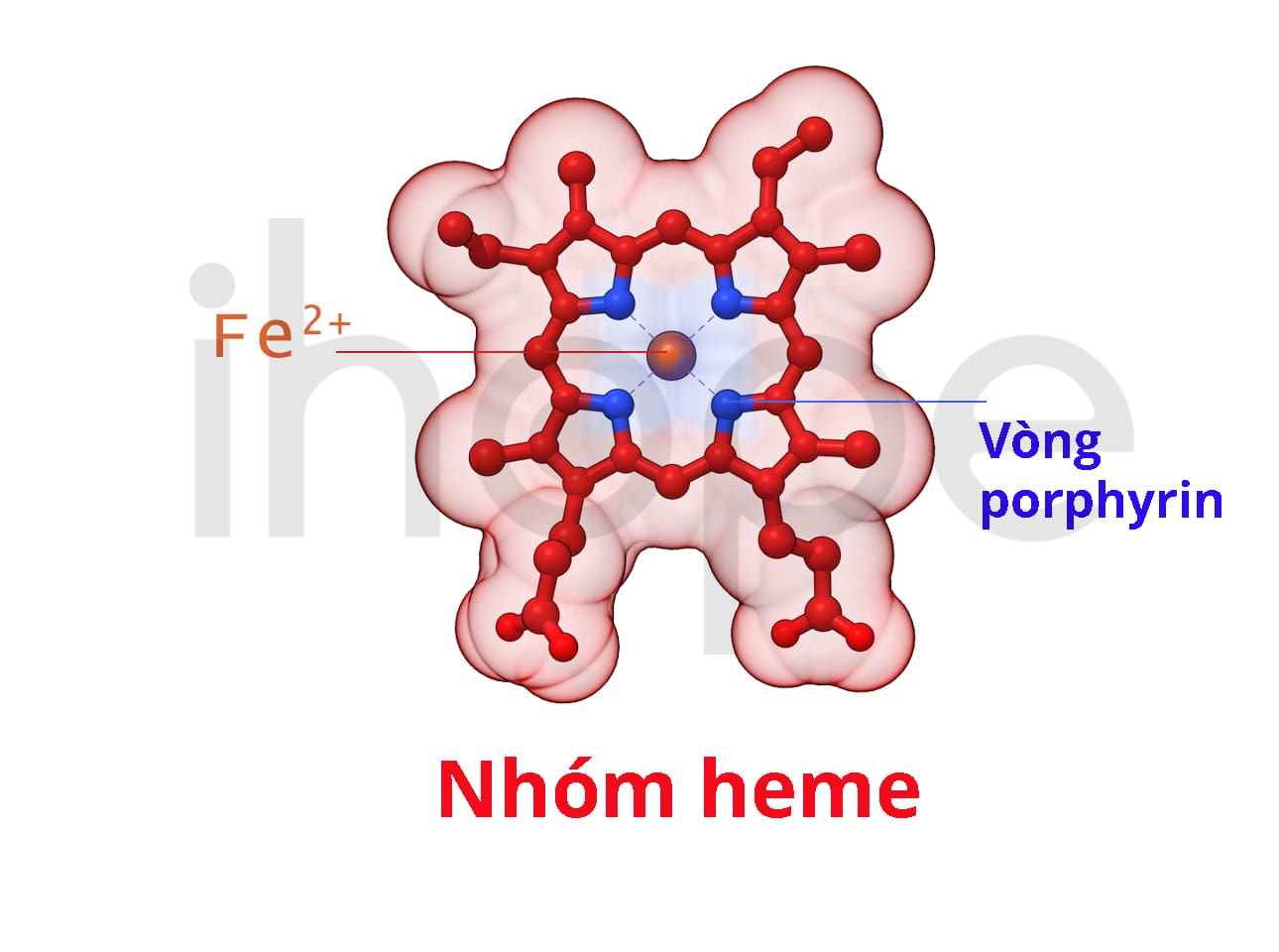
Nguồn: biologicmodels.com
Các loại hemoglobin trước khi sinh
Con người có tổng cộng 6 loại hemoglobin hồng cầu xuyên suốt quá trình phát triển. Thời gian xuất hiện và thành phần từng loại hemoglobin thay đổi tùy theo thời kỳ. Trong thời kỳ phôi thai có ba loại là hemoglobin Gower I, hemoglobin Gower II và hemoglobin Porland I.
Hemoglobin Gower I
Hemoglobin Gower I là hemoglobin được sản xuất trong giai đoạn phôi thai. Cấu trúc của hemoglobin Gower I bao gồm hai chuỗi ζ-globin (zeta-globin) và hai chuỗi ε-globin (epsilon-globin). Trong hai tháng đầu của thai kỳ, do thai nhi chưa có khả năng sử dụng hemoglobin A nên hemoglobin Gower I được sản xuất để đảm bảo cung cấp đủ oxy cho các tế bào và mô. Tuy nhiên, sau khi trẻ ra đời, hemoglobin Gower I sẽ bị thay thế thành hemoglobin A.
Hemoglobin Gower II
Tương tự như hemoglobin Gower I, hemoglobin Gower II cũng tồn tại trong hai tháng đầu thai kỳ và tham gia vào quá trình vận chuyển oxy trong giai đoạn phát triển của thai nhi. Cấu trúc của chúng bao gồm:
- Hai chuỗi α-globin tương tự hemoglobin A
- Hai chuỗi ε-globin khác với hemoglobin A, đây là điểm phân biệt và đặc trưng của hemoglobin sản xuất trong giai đoạn phôi thai
Sau khi trẻ ra đời, Hemoglobin Gower II sẽ được thay thành hemoglobin A trong máu người trưởng thành.
Hemoglobin Porland
Hemoglobin Porland thường xuất hiện trong phôi thai được 2–3 tuần với hàm lượng rất thấp. Chúng có chức năng tương tự như các hemoglobin khác trong giai đoạn phôi thai và sẽ bị thay thế thành hemoglobin A sau khi trẻ chào đời. Cấu trúc của hemoglobin Porland gồm hai chuỗi ζ-globin (zeta-globin) và hai chuỗi γ-globin (gamma-globin).
Các loại hemoglobin sau khi sinh
Đối với người trưởng thành, trong cơ thể có ba dạng hemoglobin là:
- Hemoglobin A phổ biến nhất, chiếm tỉ lệ 95–98%, chúng gồm hai chuỗi hai chuỗi α-globin (alpha-globin) và hai chuỗi β-globin (beta-globin).
- Hemoglobin A2 chiếm tỉ lệ 2–3%, chúng gồm hai chuỗi hai chuỗi α-globin (alpha-globin) và hai chuỗi δ-globin (delta-globin).
- Hemoglobin F chiếm tỉ lệ ít nhất, dưới 1%, chúng chứa hai chuỗi α-globin (alpha-globin) và hai chuỗi γ-globin (gamma-globin).
Xem thêm: Xem thêm Hemoglobin F
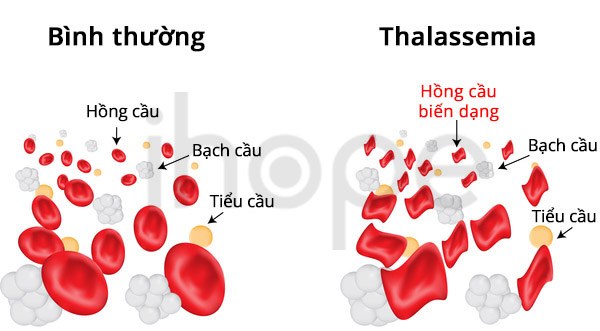
Mỗi loại hemoglobin có một tỷ lệ và chuỗi globin nhất định, nếu tỷ lệ này thay đổi sẽ xuất hiện bệnh lý. Chuỗi globin giảm số lượng dẫn đến bệnh thalassemia trong khi chuỗi globin bị thay đổi cấu trúc, trật tự axit amin sẽ tạo ra các loại hemoglobin bất thường như
Hemoglobin S (HbS hoặc hồng cầu hình liềm)
Hemoglobin S có chuỗi polypeptide của β-globin khác với hemoglobin bình thường. Cụ thể, đối với hemoglobin S tại vị trí axit amin thứ 6 trong chuỗi β-globin, glutamic được thay thế bằng valine. Điều này dẫn đến hemoglobin S bị thay đổi chức năng. Khi oxyhemoglobin giải phóng oxy ra khỏi heme, hemoglobin S sẽ hình thành tế bào hông cầu hình liềm (sickle cells). Cấu trúc hình liềm có dạng góc cạnh, chúng có thể gây tắc những nghẽn mạch máu nhỏ, giảm khả năng vận chuyển oxy của tế bào hồng cầu, từ đó gây ra bệnh thiếu máu hồng cầu hình liềm.
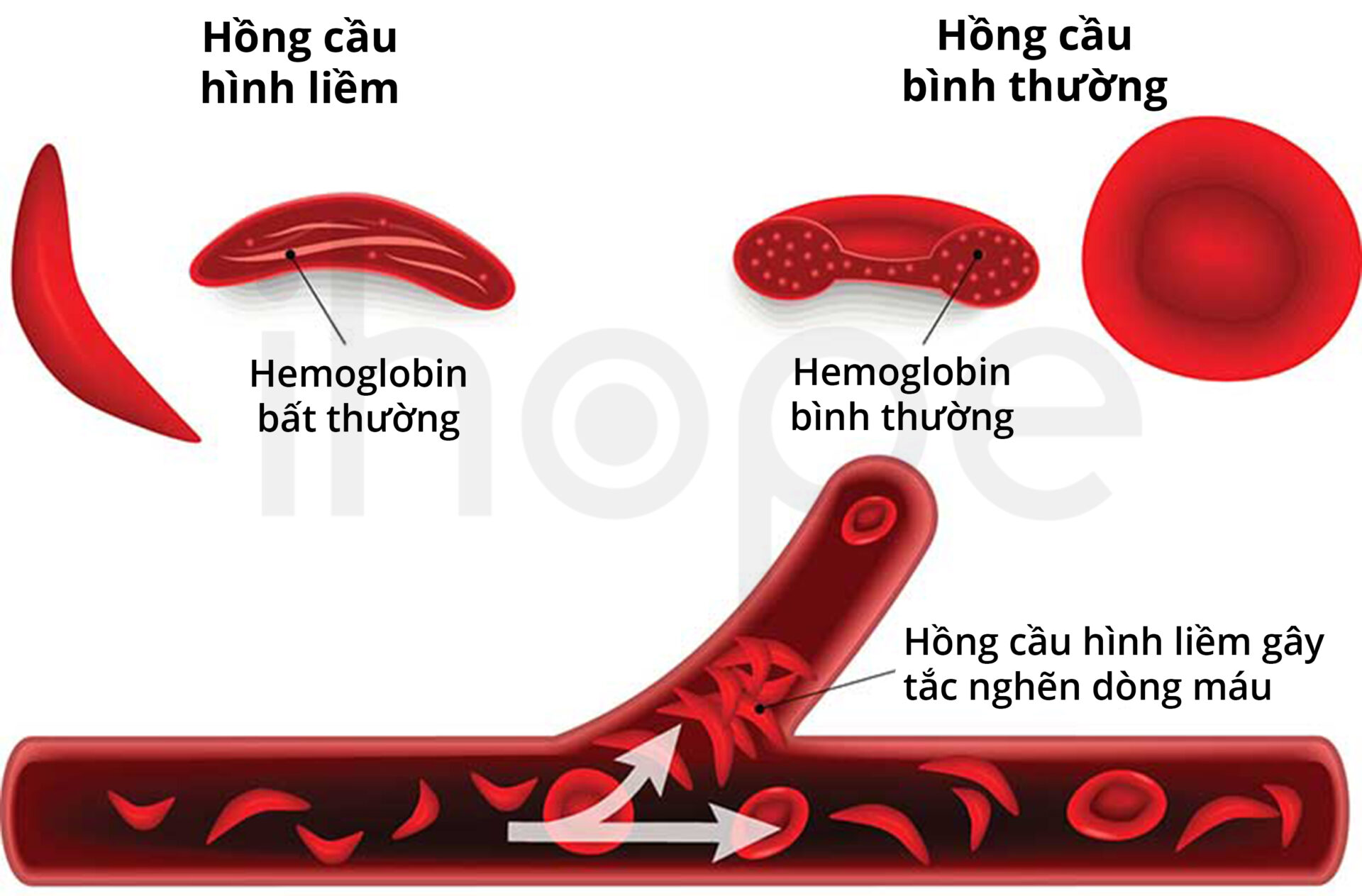
Nguồn: Froedtert & MEDICAL COLLEGE of WISCONSIN
Thiếu máu hồng cầu hình liềm là bệnh di truyền gây ra nhiều vấn đề sức khỏe như khó thở, thiếu máu và mệt mỏi. Các cơ quan và mô khác trong cơ thể như não , gan , thận cũng bị ảnh hưởng nghiêm trọng do thiếu oxy. Bên cạnh đó, hệ miễn dịch bị suy yếu khiến người bệnh có thể tăng khả năng mắc nhiều bệnh nhiễm trùng khác. Bệnh di truyền theo kiểu lặn trên nhiễm sắc thể thường, nghĩa là bệnh chỉ biểu hiện khi có cả hai bản sao của gen đột biến trong mỗi tế bào. Người mang một bản sao của gen hemoglobin S và một bản sao của gen hemoglobin bình thường (HbA) thường không bị bệnh, nhưng có thể xuất hiện triệu chứng nhẹ.

Nguồn: U.S. National Library of Medicine
Xem thêm: Xem thêm Hồng cầu hình liềm
Hemoglobin E (HbE)
Hemoglobin E (HbE) có chức năng vận chuyển oxy từ phổi đến các tế bào và mô trong cơ thể. Hemoglobin E rất phổ biến tại khu vực Đông Nam Á như Thái Lan, Campuchia, Lào và Việt Nam. Hemoglobin E có chuỗi polypeptide của β-globin khác với hemoglobin bình thường. Cụ thể, tại vị trí axit amin thứ 26 trong chuỗi β-globin, lysine được thay thế bằng glutamic.
Hemoglobin E di truyền lặn, nên bệnh chỉ biểu hiện khi có cả hai bản sao của gen đột biến trong mỗi tế bào. Tuy nhiên, hemoglobin E thường không được xem là bệnh, nhiều người mang hai bản sao hemoglobin E có cuộc sống hoàn toàn bình thường. Trong một số trường hợp, người bệnh có thể trải qua những triệu chứng như thiếu máu, mệt mỏi và thiếu sắt nhưng không nghiêm trọng.
Thông thường, hemoglobin E chỉ gây ra bệnh khi kết hợp với biến thể hemoglobin khác như:
- Bệnh HbAE Bart’s do sự kết hợp giữa hemoglobin A, hemoglobin E và hemoglobin Bart
- Bệnh HbEF Bart’s do sự hiện diện của hemoglobin E, hemoglobin F và hemoglobin Bart
Xem thêm: Xem thêm Bệnh Hemoglobin E
Hemoglobin C (HbC)
Tương tự như hemoglobin S, hemoglobin C có chuỗi polypeptide của β-globin khác với hemoglobin bình thường. Cụ thể, đối với hemoglobin C tại vị trí axit amin thứ 6 trong chuỗi β-globin, glutamic được thay thế bằng lysine. Do đó, hemoglobin C giảm khả năng hòa tan trong hồng cầu, dẫn đến hình thành tinh thể, từ đó làm tăng độ nhớt của máu và giảm tuổi thọ hồng cầu.
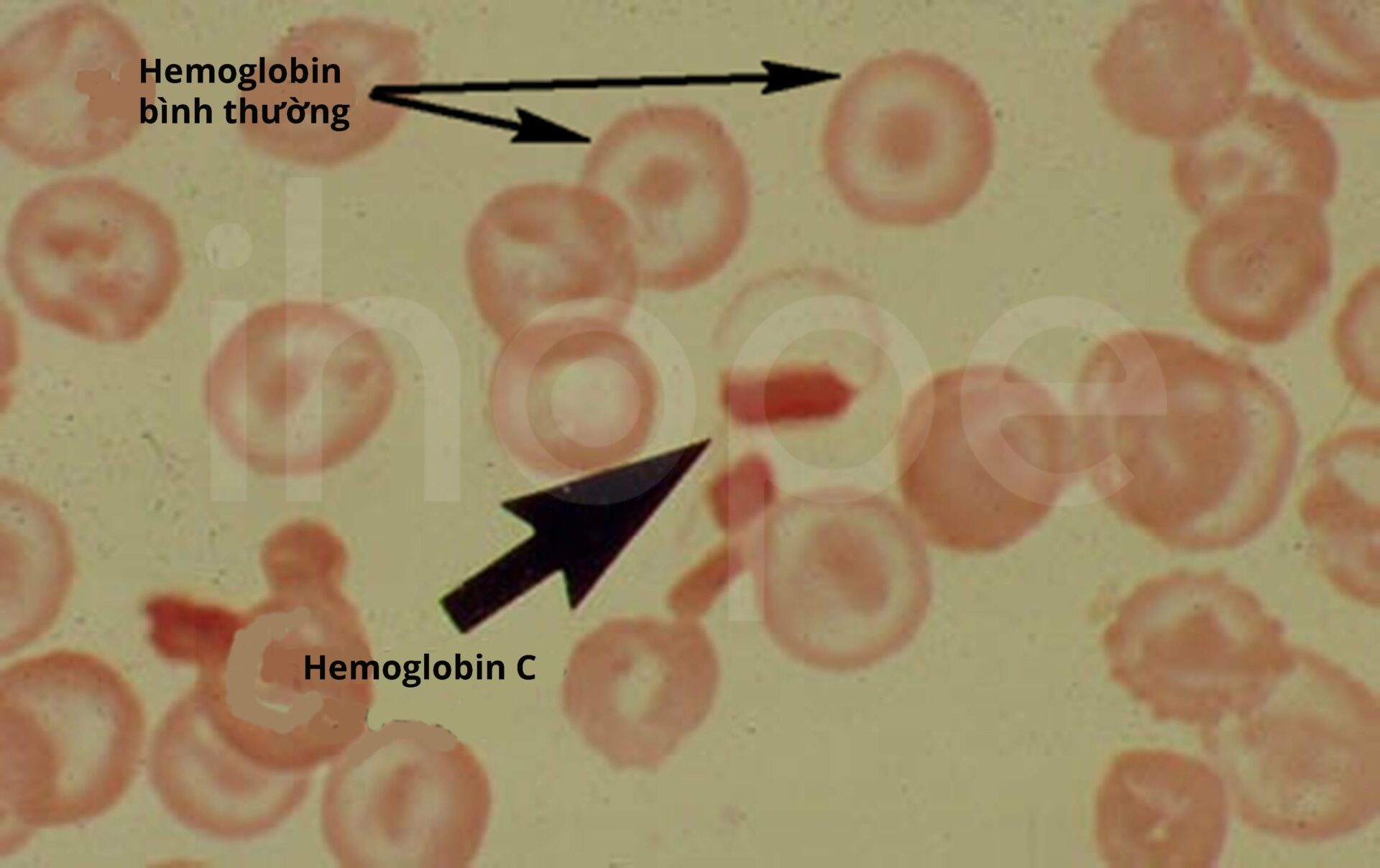
Nguồn: Medical Labs
Đa phần hemoglobin C không gây ra bệnh nhưng một số trường hợp, người bệnh có biểu hiện thiếu máu tán huyết dạng nhẹ bao gồm:
- Mệt mỏi
- Chóng mặt
- Lách to
- Vàng da
Tuy nhiên, khác với bệnh hồng cầu hình liềm, hemoglobin C không gây tắc mạch máu trừ khi chúng kết hợp với hemoglobin S (Hb SC). Đối với bệnh nhân bị tan máu mãn tính, họ có thể phát triển thành bệnh sỏi mật sắc tố. Nếu bệnh nhân mắc bệnh hồng cầu hình liềm kết hợp hemoglobin C, họ có nguy cơ bị tắc tĩnh mạch võng mạc, hoại tử chỏm xương đùi do thiếu máu và huyết khối tại vi mạch tủy thận.
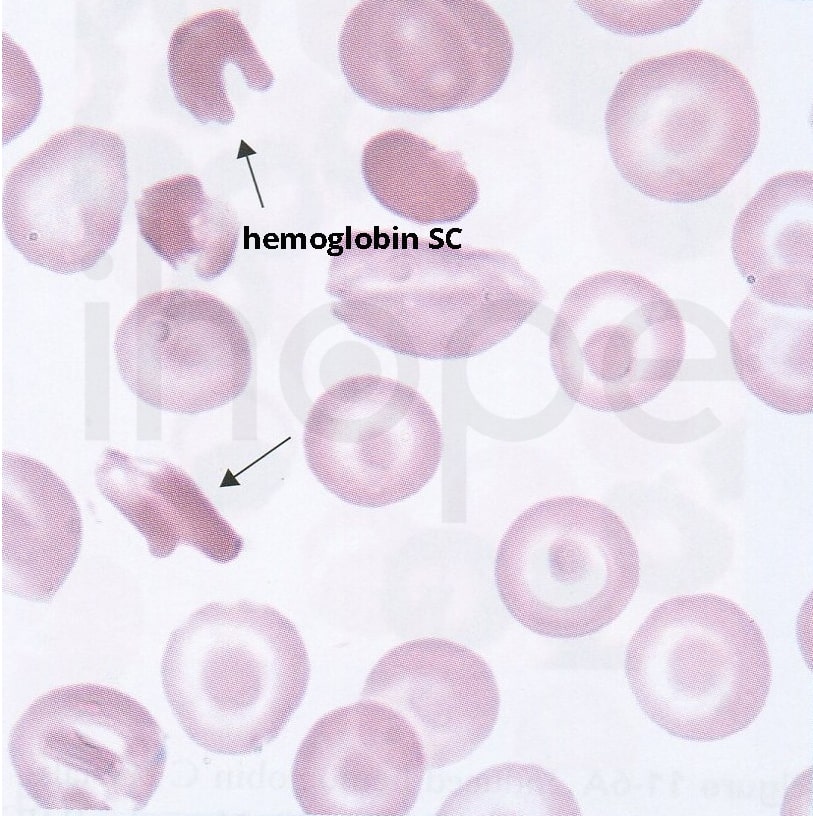
Nguồn: Medical Labs
Hemoglobin C di truyền theo kiểu lặn, nên bệnh chỉ biểu hiện khi có cả hai bản sao của gen đột biến trong mỗi tế bào. Bên cạnh đó, hemoglobin C có khả năng kết hợp với các hemoglobin khác như hemoglobin S, hemoglobin C và hemoglobin E để gây ra những bệnh lý khác.
Hemoglobin D (HbD)
Hemoglobin D có chuỗi polypeptide của β-globin khác với hemoglobin bình thường. Cụ thể, tại vị trí axit amin thứ 121 trong chuỗi β-globin, axit glutamic được thay thế bằng glutamine. Hemoglobin D phổ biến tại quần thể người gốc Ấn, Châu Á, Châu Âu, đặc biệt người Anh và Ireland.
Bốn loại hemoglobin D thường gặp bao gồm:
- Hemoglobin D đặc trưng: trẻ thừa hưởng một hemoglobin D từ bố hoặc mẹ và một hemoglobin A từ người còn lại. Dạng này thường không ảnh hưởng đến sức khỏe.
- Hemoglobin D dạng đồng hợp: trẻ thừa hưởng hai hemoglobin D từ bố và mẹ. Trẻ thuộc dạng này có thể bị thiếu máu tán huyết nhẹ trong vài tháng đầu đời khi lượng hemoglobin thai nhi giảm trong khi hemoglobin D tăng lên.
- Hemoglobin D hồng cầu hình liềm: trẻ thừa hưởng một hemoglobin D từ bố hoặc mẹ và một hemoglobin S từ người còn lại. Trẻ bị thiếu máu tán huyết từ nhẹ đến trung bình trong vài tháng đầu đời khi lượng hemoglobin thai nhi giảm và lượng hemoglobin D, hemoglobin S tăng lên. Người bệnh thuộc dạng này ít gặp nhiễm trùng liên quan đến lá lách cũng như các cơ quan khác chịu ảnh hưởng nhẹ hơn so với bệnh hồng cầu hình liềm.
- Hemoglobin D/β Thalassemia: trẻ bị thiếu máu tán huyết từ nhẹ đến trung bình, tùy thuộc vào chuỗi β-globin bị ảnh hưởng. Trẻ có lượng hemoglobin giảm, hemoglobin D tăng, lách to và thiếu sắt.
Nguồn: joshya/Shutterstock.com
Các loại hemoglobin hiếm gặp
Hemoglobin M
Đa phần hemoglobin M hình thành do sự thay thế axit amin tyrosine thành histidine trong chuỗi α, β hoặc γ-globin. Mỗi chuỗi globin tương ứng với biến thể khác nhau:
- Biến thể do thay thế axit amin trong chuỗi α-globin: hemoglobin M-Boston, hemoglobin M-Iwate và hemoglobin Auckland
- Biến thể do thay thế axit amin trong chuỗi β-globin: hemoglobin Chile, hemoglobin M-Saskatoon, hemoglobin M-Milwaukee-1 và hemoglobin M-Milwaukee-2
- Biến thể do thay thế axit amin trong chuỗi γ-globin: hemoglobin FM-Osaka và hemoglobin FM-Fort Ripley
Thay đổi này khiến nhân heme oxy hóa thành dạng methemoglobin (ion sắt Fe3+) dẫn đến hemoglobin không có khả năng vận chuyển oxy. Do đó, da người bệnh tím tái và máu màu nâu sẫm. Các biến thể hemoglobin M khác nhau có thể gây ra dấu hiệu và triệu chứng khác nhau.
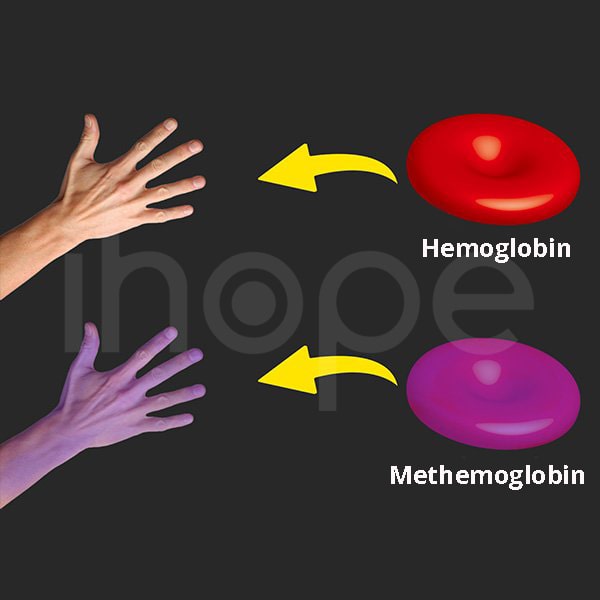
Biokiduniya.com
Hemoglobin Bassett
Hemoglobin Bassett có chuỗi polypeptide của α-globin khác với hemoglobin bình thường. Cụ thể, tại vị trí axit amin thứ 94 trong chuỗi α-globin, aspartic được thay thế bằng alanine. Do thay đổi cấu trúc, hemoglobin Bassett giảm ái lực với oxy.
Hemoglobin O-Arab
Hemoglobin O-Arab có chuỗi polypeptide của β-globin khác với hemoglobin bình thường. Cụ thể, tại vị trí axit amin thứ 121 trong chuỗi β-globin, glutamic được thay thế bằng lysine. Khi kết hợp với hemoglobin S, chúng gây ra hemoglobin S/O-Arab—một dạng bệnh hồng cầu hình liềm nghiêm trọng.
Hemoglobin Lepore
Cấu trúc của hemoglobin Lepore bao gồm hai chuỗi α-globin bình thường và hai chuỗi βδ-globin (beta-delta globin) bất thường. Chuỗi βδ-globin được tạo thành do sự kết hợp giữa locus gen β-globin (HBB) và locus gen δ-globin (HBD) trong quá trình giảm phân. Lượng hemoglobin Lepore hiện diện trong máu người bệnh dao động từ 8–30%, phần còn lại là hemoglobin F với số lượng rất nhỏ (thường dưới 1%).
Hemoglobin Lepore di truyền theo kiểu lặn và có triệu chứng tương tự bệnh beta-thalassemia. Trong 5 năm đầu đời, người bệnh có xu hướng thiếu máu nghiêm trọng. Họ cũng biểu hiện một số triệu chứng như gan to, lách to, tim to và bất thường xương .
Xem thêm: Beta thalassemia
Hemoglobin Hope
Hemoglobin Hope có chuỗi polypeptide của β-globin khác với hemoglobin bình thường. Cụ thể, tại vị trí axit amin thứ 136 trong chuỗi β-globin, glycine được thay thế bằng aspartic. Do đó, hemoglobin giảm ổn định cấu trúc mức độ nhẹ và giảm ái lực với oxy. Nhìn chung, hemoglobin Hope không ảnh hưởng nhiều đến người bệnh.
Hemoglobin J
Hemoglobin J có chuỗi polypeptide của α-globin khác với hemoglobin bình thường. Cụ thể, tại vị trí axit amin thứ 120 trong chuỗi α-globin, alanine được thay thế bằng glutamic. Người có hemoglobin J phần lớn không có biểu hiện thiếu máu.
Hemoglobin N-Baltimore
Hemoglobin N-Baltimore có chuỗi polypeptide của β-globin khác với hemoglobin bình thường. Cụ thể, tại vị trí axit amin thứ 95 trong chuỗi β-globin, lysine được thay thế bằng glutamic.
Hemoglobin Louisville
Hemoglobin Louisville có độ ổn định cấu trúc thấp do gốc phenylalanyl tại vị trí 42 được thay thế bằng leucyl. Bên cạnh đó, hemoglobin Louisville cũng giảm ái lực với oxy và giảm tương tác giữa các nhân heme.
Hemoglobin Vanvitelli
Hemoglobin Vanvitelli có chuỗi polypeptide của α-globin khác với hemoglobin bình thường. Cụ thể, tại vị trí axit amin thứ 43 trong chuỗi α-globin, phenylalanine được thay thế bằng leucine. Hemoglobin Vanvitelli có thể gây ra nhiều triệu chứng khác nhau như chứng da xanh tím, khó thở, thiếu máu tán huyết mãn tính.

Nguồn: Cleveland Clinic
Lời kết
Hemoglobin là thành phần quan trọng của hệ thống tuần hoàn máu trong cơ thể. Hemoglobin cũng đại diện cho khía cạnh di truyền phức tạp của con người. Những biến thể của hemoglobin như hemoglobin S, hemoglobin C, hemoglobin E và hemoglobin D có thể gây ra nhiều vấn đề sức khỏe. Do đó, hiểu rõ về cấu trúc và mức độ ảnh hưởng của các biến thể này có thể giúp chẩn đoán và điều trị tốt hơn bệnh lý liên quan đến hemoglobin.
Hiện nay, những biến thể hemoglobin có thể được chẩn đoán thông qua sàng lọc sơ sinh. Trong vòng 24–48 giờ sau khi sinh, trẻ được lấy máu gót chân để thực hiện sàng lọc sơ sinh nhằm phát hiện các bất thường hemoglobin và các rối loạn khác. Nếu kết quả cho thấy nguy cơ cao mắc bệnh, bác sĩ sẽ cho em bé làm thêm xét nghiệm chẩn đoán. Điều quan trọng cần lưu ý là kết quả sàng lọc nguy cơ cao không đồng nghĩa với kết luận em bé mắc bệnh. Kết quả này có thể do mẫu máu ban đầu được thu quá ít hoặc quá sớm. Tuy nhiên, cha mẹ nên đưa trẻ tái khám để làm xét nghiệm xác nhận. Nếu không được điều trị, bệnh có thể ảnh hưởng đến sức khỏe của trẻ rõ rệt ngay sau khi sinh, xét nghiệm tiếp theo phải được tiến hành càng sớm càng tốt để xác định xem liệu trẻ có mắc bệnh hay không.
Xem thêm: Các loại xét nghiệm sàng lọc sơ sinh
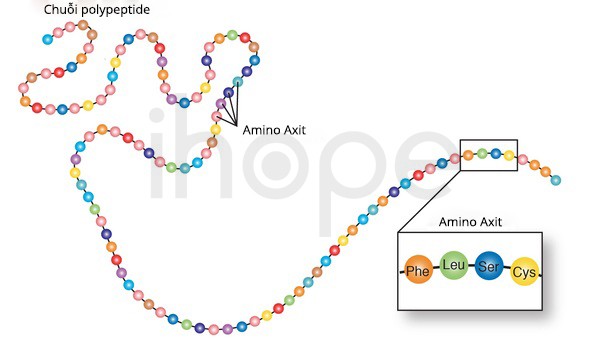
Ảnh: Axit amin
Nguồn: Darryl Leja, NHGRI
Ảnh: Cấu trúc bên trong của não
Nguồn: Terese Winslow
Ảnh: giải phẫu gan người
Nguồn: Terese Winslow LLC
Ảnh: Thận và tuyến thượng thận
Nguồn: National Cancer Institue
Ảnh: Cấu trúc tim
Nguồn: American Heart Association
Ảnh: Giải phẫu xương người
Nguồn: Terese Winslow LLC
References
- Centers for Disease Control and Prevention What is Sickle Cell Disease? Retrieved October 25, 2023 from https://www.cdc.gov/ncbddd/sicklecell/facts.html
- National Institute of Health. Hemoglobin: Structure, Function and Allostery. Retrieved October 25, 2023 from https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7370311/
- National Institute of Health. Structure-function relations of human hemoglobins. Retrieved October 25, 2023 from https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1484532/
- National Institute of Health. Human embryonic, fetal, and adult hemoglobins have different subunit interface strengths. Correlation with lifespan in the red cell. Retrieved October 25, 2023 from https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2203358/
- National Institute of Health. Hemoglobin C Disease. Retrieved October 25, 2023 from https://www.ncbi.nlm.nih.gov/books/NBK559043/
- National Institute of Health. The Hemoglobin E Thalassemias. Retrieved October 25, 2023 from https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3405827/
- National Institute of Health. Hemoglobin M disease. Retrieved October 25, 2023 from https://www.ncbi.nlm.nih.gov/medgen/777099
- National Institute of Health. Characterization of hemoglobin bassett (alpha94Asp-->Ala), a variant with very low oxygen affinity. Retrieved October 25, 2023 from https://pubmed.ncbi.nlm.nih.gov/15495251/
- National Institute of Health. Hemoglobin O Arab in interaction with beta 0-thalassemia. Retrieved October 25, 2023 from https://pubmed.ncbi.nlm.nih.gov/2739498/
- National Institute of Health. Homozygous Hemoglobin Lepore: A Rare Condition Seen in a Bangladeshi Family. Retrieved October 25, 2023 from https://pubmed.ncbi.nlm.nih.gov/34605493/
- National Institute of Health. Hemoglobin Variants: Biochemical Properties and Clinical Correlates. Retrieved October 25, 2023 from https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3579210/
- Orphanet. Hemoglobin D disease. Retrieved October 25, 2023 from https://www.orpha.net/consor/cgi-bin/OC_Exp.php?lng=en&Expert=90039