Hemoglobin A hay HbA không phải bệnh, nó là huyết sắc tố tồn tại trong máu của người trưởng thành khỏe mạnh. Chức năng chính của HbA là vận chuyển oxy đến tế bào. Ngoài ra, HbA còn tham gia xúc tác nhiều phản ứng sinh hóa khác trong cơ thể, chuyển hóa oxit nitric, định hướng quá trình trao đổi chất, điều chỉnh pH và duy trì cân bằng oxy hóa khử.
Cấu trúc và chức năng
Cấu trúc HbA
Trong phần lớn động vật có xương sống (kể cả con người), hemoglobin được hình thành từ 4 phần nhỏ (tetramer). Hai tiểu phần chính của hemoglobin bao gồm hai tiểu đơn vị α (α1 và α2 ) và hai tiểu đơn vị β (β1 và β2 ); các tiểu đơn vị này gip61ng nhau về cấu trúc và kích thước. Thông thường, mỗi tiểu đơn vị liên kết với một nhân heme. Heme bao gồm một vòng porphyrin có khả năng tạo phức với sắt (dạng Fe2+). Heme là nhân tố có khả năng gắn kết với oxy và tạo màu đỏ của hồng cầu. Một phân tử hemoglobin có thể liên kết với 4 phân tử oxy.
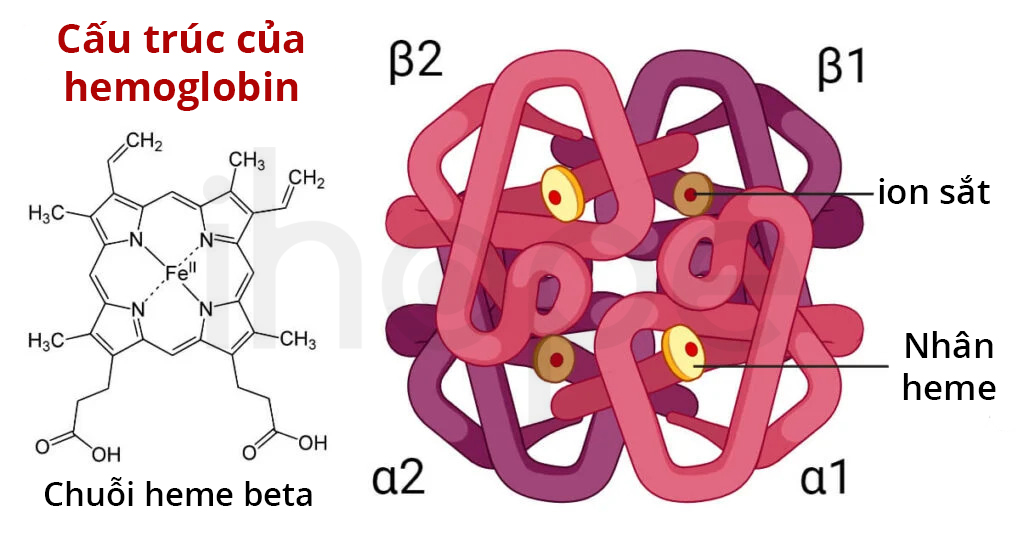
Nguồn: Microbe Notes
Chức năng HbA
Chức năng chính của hemoglobin là trao đổi và vận chuyển oxy trong cơ thể.
Hemoglobin có 2 trạng thái cấu trúc bậc 4, bao gồm:
- Dạng deoxy (ái lực với oxy thấp)
- Dạng oxy (ái lực với oxy cao)
Những biến đổi trong cấu trúc bậc 4 của phân tử hemoglobin dẫn đến thay đổi ái lực với oxy. Môi trường là một trong những yếu tố quyết định. Ví dụ, trong môi trường vi mô của phổi, các điều kiện thích hợp cho trạng thái ái lực cao với oxy, do đó, hemoglobin có khả năng liên kết với oxy. Khi di chuyển đến các mô và cơ quan, hemoglobin giảm ái lực với oxy nên nó giải phóng oxy vào tế bào.
Các bệnh liên quan đến hemoglobin A
Hơn 1000 biến thể hemoglobin xảy ra tự nhiên đã được ghi nhận. Những đột biến này thay đổi khả năng liên kết với oxy của HbA. Một số bất thường đáng chú ý gây ra các bệnh có mức độ từ nặng đến nhẹ bao gồm đa hồng cầu, methemoglobin huyết, hội chứng da xanh tím, thiếu oxy mô và chứng suy hô hấp.
Alpha thalassemia
Alpha thalassemia hay còn gọi là bệnh thiếu máu tán huyết thể alpha. Nguyên nhân gây ra bệnh do bất thường quá trình sản xuất chuỗi alpha–globin từ hai gen HBA1 và HBA2. Thông thường, tế bào có 2 bản sao gen HBA1 và 2 bản sao gen HBA2. Đột biến mất một vài trong số các bản sao này dẫn đến các dạng khác nhau của alpha thalassemia.
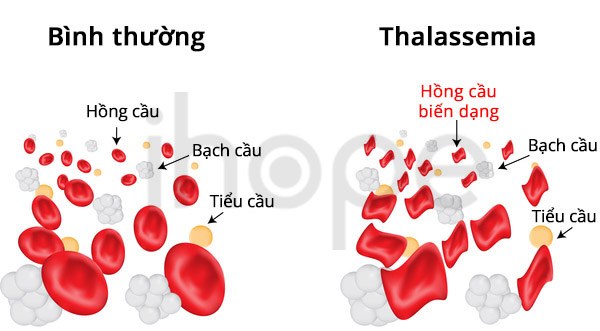
Nguồn: joshya/Shutterstock.com
Những biểu hiện phổ biến của bệnh alpha thalassemia có thể bao gồm:
- Mệt mỏi, mất sức hoặc hụt hơi
- Dáng vẻ nhợt nhạt, vàng da, vàng mắt
- Dễ cáu gắt
- Dị dạng xương mặt
- Phát triển chậm
- Bụng phình to
- Nước tiểu sẫm màu
Hội chứng Hb Bart’s hay hội chứng phù thai nhi Hb Bart’s là bệnh alpha thalassemia thể nghiêm trọng với biểu hiện phù thai. Em bé thường bị thiếu máu, gan lách to, dị tật tim, hệ tiết niệu và sinh sản bất thường. Thai nhi thường tử vong trong bụng mẹ hoặc ngay sau sinh.
Bệnh HbH là dạng nhẹ của alpha thalassemia. Người bệnh thường bị thiếu hồng cầu nhẹ, gan và lách to, vàng da hoặc mắt.
Alpha thalassemia có dạng di truyền phức tạp. Nguy cơ mắc bệnh phụ thuộc vào số lượng bản sao bị mất và sự kết hợp giữa gen HBA1 và HBA2. Trường hợp cả cha và mẹ đều mang đột biến mất một bản sao, khả năng cao con mắc hội chứng Hb Bart’s hoặc HbH.
Xem thêm: Xem thêm Alpha Thalassemia
Beta thalassemia
Tương tự thiếu máu tán huyết thể alpha, beta thalassemia xảy ra do quá trình sản xuất chuỗi beta–globin trong HbA bị bất thường. Đột biến gen HBB khiến số lượng beta–globin suy giảm, nên tế bào máu không thể trưởng thành để thực hiện chức năng.
Trẻ mắc bệnh beta thalassemia thể nặng (chứng thiếu máu cooley) khởi phát trong 2 năm đầu sau sinh. Bệnh nhi thường chậm phát triển, vàng da và mắt, gan lách to, dị tật xương hoặc tim. Đối với beta thalassemia thể vừa, trẻ có biểu hiện thiếu máu nhẹ đến trung bình và có thể kèm theo một số vấn đề sức khỏe khác.
Bệnh do đột biến gen HBB di truyền lặn trên nhiễm sắc thể thường. Phần lớn bệnh nhân có cả bố và mẹ đều mang gen đột biến, nhưng bố mẹ không có biểu hiện bất thường. Một số trường hợp mang đột biến một bản sao gen HBB bị thiếu máu nhẹ.
Xem thêm: Xem thêm Beta Thalassemia
Hồng cầu hình liềm
Hồng cầu hình liềm (sickle cell disease) là biến thể phổ biến nhất của hemoglobin. Bất thường xảy ra khi có đột biến điểm (βGlu6Val) trong chuỗi β–globin của hemoglobin bình thường. Đột biến khiến các phân tử trong hemoglobin liên kết với nhau thành chuỗi polyme không hòa tan, nên tế bào hồng cầu có hình liềm thay vì dạng đĩa tròn. Do đó, tế bào máu mất đi tính linh hoạt, chúng dính vào nhau rồi cản trở lưu lượng máu và quá trình trao đổi oxy trong cơ thể. Ngoài ra, hồng cầu hình liềm thường kém bền, nên cơ thể bị thiếu máu.
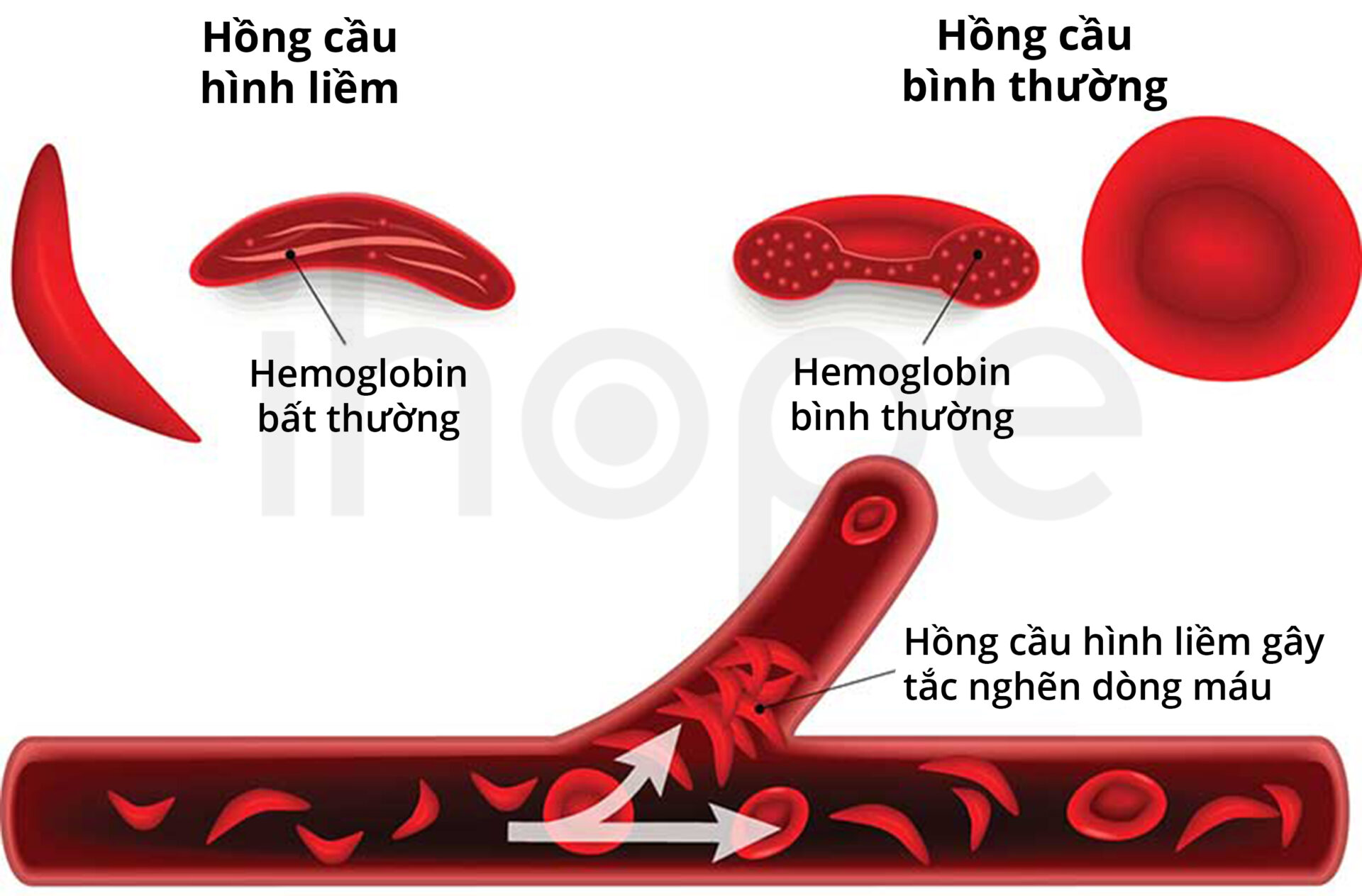
Nguồn: Froedtert & MEDICAL COLLEGE of WISCONSIN
Có nhiều dạng hồng cầu hình liềm tùy thuộc vào kiểu gen đột biến:
- Hemoglobin SS (HbSS): bệnh thể nặng, chiếm khoảng 65% số trường hợp hồng cầu hình liềm. Bệnh nhân thừa hưởng gen tạo ra hemoglobin S từ bố và mẹ. HbSS gây ra thiếu máu mãn tính do phần lớn hoặc toàn bộ hemoglobin của người bệnh đều bất thường.
- Hemoglobin SC (HbSC): bệnh có mức độ từ nhẹ đến trung bình. Khoảng 25% số trường hợp hồng cầu hình liềm thuộc dạng này. Bệnh nhân thừa hưởng một bản sao gen hemoglobin S từ cha hoặc mẹ.
- Hemoglobin (HbS) beta thalassemia: bệnh nhân thừa hưởng một gen hemoglobin S từ cha (hoặc mẹ) và một bản sao gen đột biến gây bệnh beta thalassemia từ người còn lại. Bệnh có hai loại bao gồm HbS beta+ với các biểu hiện nhẹ và HbS beta 0 có triệu chứng nghiêm trọng.
- Các loại khác (HbSD, HbSE, HbSO): bệnh gây ra bởi một bản sao gen hemoglobin S và một bản sao gen đột biến khác (D,E hoặc O).
Xem thêm: Xem thêm Hồng cầu hình liềm
Methemoglobin huyết
Methemoglobin huyết (MetHb) là bệnh về máu khiến các hemoglobin có thể vận chuyển oxy nhưng mất khả năng giải phóng oxy vào tế bào và mô hiệu quả. Nguyên nhân thường do sử dụng một số loại thuốc kích thích hoặc tiếp xúc với hóa chất và có thể do đột biến gen.
Đối với methemoglobin huyết do đột biến gen, bệnh di truyền theo kiểu trội hoặc lặn trên nhiễm sắc thể thường.
Methemoglobin huyết di truyền lặn
Đột biến gen CYB5R gây ra bệnh dạng lặn trên nhiễm sắc thể thường. Gen CYB5R cung cấp hướng dẫn tạo ra enzyme cytochrome b5 reductase 3. Enzyme này chuyển các electron mang điện tích âm từ phân tử này sang phân tử khác. Thông thường, ion sắt trong hemoglobin là Fe2+ có khả năng liên kết với oxy. Tuy nhiên, ion này có thể tự chuyển thành dạng Fe3+ và không gắn được oxy (dạng methemoglobin). Dạng methemoglobin chiếm 2% trong tế bào hồng cầu. Enzyme cytochrome b5 reductase 3 tham gia vào quá trình chuyển đổi Fe3+ thành Fe2+ để homoglobin tiếp tục cung cấp oxy cho cơ thể.
Đột biến gen CYB5R gây ra methemoglobin di truyền lặn trên nhiễm sắc thể thường khiến enzyme cytochrome b5 reductase 3 mất chức năng. Từ đó, tỉ lệ methemoglobin tăng lên từ 10 đến 50%.
Methemoglobin huyết có 2 loại bao gồm:
- Loại 1: còn gọi là thiếu men hồng cầu reductase (erythrocyte reductase deficiency)
- Loại 2: chứng thiếu men reductase tổng hợp
Bệnh chỉ biểu hiện khi có cả hai bản sao của gen đột biến trong mỗi tế bào. Người mắc bệnh lặn trên nhiễm sắc thể thường có bố và mẹ mang một bản sao của gen đột biến, nhưng bố mẹ ít khi biểu hiện triệu chứng bệnh.

Nguồn: U.S. National Library of Medicine
Methemoglobin huyết di truyền trội
Methemoglobin huyết di truyền trội trên nhiễm sắc thể thường còn được gọi là bệnh hemoglobin M (HbM). Nguyên nhân do những khiếm khuyết trong protein huyết sắc tố được tạo ra từ gen hemoglobin M đột biến. Mỗi tế bào chỉ cần một bản sao gen đột biến để biểu hiện bất thường.

Nguồn: U.S. National Library of Medicine
Chứng da xanh tím
Chứng da xanh tím (cyanosis) khiến da tại những vùng như đầu ngón tay, ngón chân và móng có màu xanh tím bất thường. Một số trường hợp nghiêm trọng, môi và nướu cũng có thể chuyển màu xanh.

Nguồn: Cleveland Clinic
Tùy thuộc vào nguyên nhân gây bệnh, các biểu hiện có thể bao gồm:
- Hạ thân nhiệt
- Tê hoặc ngứa ran tay và chân
- Ho, khó thở, thở khò khè
- Chóng mặt, mệt mỏi
Nguyên nhân do tế bào máu không nhận đủ oxy. Thông thường, máu nhận đủ oxy có màu đỏ tươi rồi lưu thông khắp cơ thể, nên da có màu hồng. Khi lưu lượng oxy không đủ, máu sẫm màu hơn nên da trở thành xanh hoặc tím. Một số bất thường trong hemoglobin khiến tế bào hồng cầu không tiếp nhận đủ oxy dẫn đến chứng xanh tím.
Thiếu oxy mô
Thiếu oxy mô (tissue hypoxia) là bất thường khiến lượng oxy trong cấp độ mô không đủ để duy trì cân bằng nội môi. Nguyên nhân chính do lưu lượng máu đến mô thấp hoặc hàm lượng oxy trong máu thấp. Các bất thường hemoglobin khiến hồng cầu giảm khả năng vận chuyển oxy đến mô gây ra các vấn đề sức khỏe.
Một số biểu hiện thường gặp của chứng thiếu oxy mô bao gồm:
- Khó thở, thở nhanh
- Rối loạn nhịp tim, tiếng thở rít
- Chức năng các cơ quan giảm
Phát hiện và chẩn đoán bệnh
Những bất thường trong HbA dẫn đến nhiều bệnh về máu, nên sức khỏe của trẻ bị ảnh hưởng nghiêm trọng. Nếu được chẩn đoán và điều trị sớm, người bệnh có thể cải thiện phần lớn các triệu chứng. Tuy nhiên, biểu hiện bệnh thường chưa rõ ràng khi trẻ sinh ra, nên bệnh thường không được phát hiện hoặc chẩn đoán chính xác. Do đó, quá trình can thiệp chậm trễ làm giảm hiệu quả điều trị.
Hiện nay, tất cả trẻ đều được khuyến nghị thực hiện xét nghiệm sàng lọc sơ sinh trong vòng một đến hai ngày sau sinh. Xét nghiệm này kiểm tra nhiều vấn đề sức khỏe hiếm gặp nhưng nghiêm trọng. Nếu kết quả sàng lọc cho thấy bất thường, bác sĩ có thể chỉ định những xét nghiệm chẩn đoán chuyên biệt hơn.
Xét nghiệm sàng lọc sơ sinh kiểm tra bao gồm 3 phần chính:
- Xét nghiệm máu: phần lớn trẻ sơ sinh được kiểm tra bằng mẫu máu. Mẫu được thu từ gót chân của trẻ (bác sĩ dùng kim chích vào gót chân của bé và thấm máu lên tờ giấy đặc biệt). Xét nghiệm này phát hiện các bất thường về hồng cầu, chuyển hóa, nội tiết và một số bệnh lý khác.
- Sàng lọc thính giác: kiểm tra bất thường hoặc mất thính lực bằng thiết bị chuyên dụng. Dựa vào kết quả xét nghiệm, bác sĩ sẽ chỉ định phương pháp can thiệp nếu cần thiết.
- Kiểm tra bất thường tim: sàng lọc dị tật tim bẩm sinh nghiêm trọng (critical congenital heart defects) thông qua đo nồng độ bão hòa oxy trong máu (pulse oximetry).
Trường hợp kết sàng lọc sơ sinh bất thường, những xét nghiệm chuyên biệt nhằm chẩn đoán chính xác bệnh bao gồm xét nghiệm di truyền, xét nghiệm hình ảnh, siêu âm.
Điều trị
Hemoglobin A có nhiều biến thể dẫn đến nhiều bệnh về máu. Ngoài ra, mức độ các triệu chứng cũng khác nhau giữa mỗi bệnh nhân dẫn đến không có phác đồ điều trị chung.
Đối với alpha và beta thalassemia, bệnh nhân có thể được truyền máu nhằm cung cấp các tế bào máu khỏe mạnh. Một số ít trường hợp bệnh nghiêm trọng đáp ứng điều trị với cấy ghép tủy sống.
Những bất thường về máu khác cần được chẩn đoán và điều trị bởi bác sĩ chuyên môn.
Kết luận
Hemoglobin A (HbA) là loại huyết sắc tố phổ biến nhất đối với người trường thành. Phân tử này giữ vai trò vận chuyển oxy đến khắp nơi trên cơ thể. Những bất thường trong HbA dẫn đến nhiều bệnh về máu và ảnh hưởng nghiêm trọng đến sức khỏe. Phát hiện và chẩn đoán bệnh sớm thông qua xét nghiệm sàng lọc sơ sinh giúp nâng cao hiệu quả điều trị.
References
- National Library of Medicine. Hemoglobin: Structure, Function and Allostery. Retrieved October 17, 2023 from https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7370311/
- U.S National Library of Medicine. Alpha thalassemia. Retrieved October 17, 2023 from https://medlineplus.gov/genetics/condition/alpha-thalassemia/
- U.S National Library of Medicine. Beta thalassemia. Retrieved October 17, 2023 from https://medlineplus.gov/genetics/condition/beta-thalassemia/
- U.S National Library of Medicine. Sickle cell disease. Retrieved October 17, 2023 from https://medlineplus.gov/genetics/condition/sickle-cell-disease/
- U.S National Library of Medicine. Autosomal recessive congenital methemoglobinemia. Retrieved October 17, 2023 from https://medlineplus.gov/genetics/condition/autosomal-recessive-congenital-methemoglobinemia/
- Cleveland Clinic. Cyanosis. Retrieved October 17, 2023 from https://my.clevelandclinic.org/health/diseases/24297-cyanosis
- KidsHealth. Newborn Screening Tests. Retrieved October 17, 2023 from https://kidshealth.org/en/parents/newborn-screening-tests.html
- National Library of Medicine. Hypoxia. Retrieved October 17, 2023 from https://www.ncbi.nlm.nih.gov/books/NBK482316/
- National Library of Medicine. Newborn Screening. Retrieved October 17, 2023 from https://www.ncbi.nlm.nih.gov/books/NBK558983/