Thiếu máu rối loạn sinh hồng cầu bẩm sinh (congenital dyserythropoietic anemia – CDA) là bệnh di truyền về máu, ảnh hưởng đến quá trình phát triển của các tế bào hồng cầu.
Nguồn: U.S National Library of Medicine
Biểu hiện lâm sàng
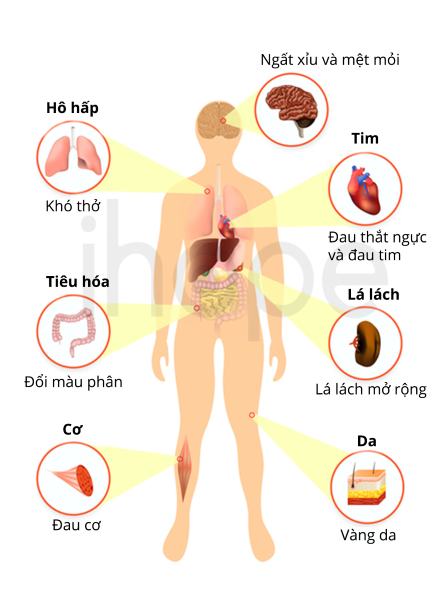
Người bệnh bị thiếu hồng cầu khỏe mạnh, nên tế bào không nhận đủ oxy. Do đó, họ luôn cảm thấy mệt mỏi, xanh xao và đau thắt ngực khi gắng sức. Các triệu chứng và thời gian khởi phát có thể khác nhau với mỗi người bệnh.
Bệnh được phân làm 4 loại như sau:
- Loại 1 thiếu máu trung bình đến nặng. Bệnh nhân thường bị vàng da, gan và lách to. Bệnh loại 1 làm dư lượng sắt quá mức gây độc cho các cơ quan. Dư lượng sắt trong thời gian dài dẫn đến loạn nhịp tim gây suy tim sung huyết. Các biến chứng nghiêm trọng hơn gồm các bệnh mãn tính như xơ gan, tiểu đường. Một số trẻ sơ sinh có dị tật xương ngón tay hoặc ngón chân. Các triệu chứng khởi phát sớm với trẻ nhỏ, tuổi dậy thì hoặc trước khi sinh.
- Loại 2 dạng phổ biến. Triệu chứng tương tự loại 1 nhưng nhẹ hơn và các biến chứng phát triển chậm.
- Loại 3 dạng hiếm nhất trong các loại và biểu hiện nhẹ nhất. Gan và lách không bị ảnh hưởng. Giảm hồng cầu lưới-huyết nhẹ và có dấu hiệu tan máu, vàng da. Người bệnh có các vấn đề về võng mạc gây suy giảm thị lực. Bệnh gammopathy đơn dòng (bệnh về máu) có thể dẫn đến bệnh bạch cầu, đây là biến chứng nghiêm trọng của loại 3.
- Loại 4 thiếu máu nghiêm trọng đe dọa tính mạng trẻ sơ sinh.
Độ phổ biến
Nhiều trường hợp mắc bệnh được ghi nhận trên toàn thế giới, trong đó loại 2 là dạng phổ biến nhất với hơn 300 ca, loại 3 và loại 4 hiếm gặp. Tỷ lệ mắc bệnh loại 1 vẫn chưa được xác định do triệu chứng bệnh dễ nhầm lẫn với một số bệnh lý khác hoặc chưa được chẩn đoán.
Nguyên nhân
Bệnh được chia thành bốn loại dựa vào nguyên nhân di truyền:
- Loại 1 do đột biến gen CDAN1 (90%) và C15RF41 (1%) gây ra. Người ta vẫn chưa hiểu rõ chức năng của các gen này. Theo nhiều nghiên cứu, gen CDAN1 cung cấp hướng dẫn tạo ra protein codanin-1 có vai trò điều hòa chu trình tế bào trong pha S. C15RF41 cung cấp hướng dẫn tạo ra một loại protein liên kết với codanin-1 để thực hiện chức năng sửa chữa và duy trì tính ổn định của ADN. Hệ thống này còn giúp tháo rời và lắp ráp các đơn vị của ADN. Đột biến một trong hai gen làm ảnh hưởng đến hệ thống sửa sai, từ đó lỗi ADN xuất hiện. Các sai hỏng này cản trở quá trình phân bào, đặc biệt các nguyên bào hồng cầu. Một số người mắc bệnh nhưng không xác định được nguyên nhân di truyền.
- Đột biến gen SEC23B gây ra bệnh loại 2. Gen SEC23B cung cấp hướng dẫn tạo ra protein Sec23B – thành phần quan trọng trong cấu trúc của các túi vận chuyển protein. Trong quá trình phát triển của tế bào hồng cầu, các túi này đảm bảo vận chuyển protein đến nơi cần thiết. Người ta vẫn đang tìm hiểu cơ chế đột biến gen SEC23B dẫn đến triệu chứng bệnh.
- Nguyên nhân di truyền của bệnh loại 3 vẫn chưa được xác định. Theo những ca bệnh được ghi nhận tại Mỹ và Thụy Điển, bệnh do đột biến gen KIF23 (15q21). Gen KIF23 cung cấp hướng dẫn tạo protein kinesin phân bào (MKLP1). Đột biến gen này ảnh hưởng đến chức năng của protein MKLP1 trong quá trình tạo ra tế bào, dẫn đến hình thành hồng cầu đa nhân lớn trong tủy xương. Các nhà nghiên cứu đang tiếp tục tìm kiếm các gen liên quan đến bệnh.
- Loại 4 do đột biến gen KLF1 gây ra. Chức năng của gen này vẫn chưa rõ ràng. Phân tích mô hình biểu hiện chung cho thấy gen đột biến tạo ra protein bất thường làm gián đoạn quá trình tạo hồng cầu, dẫn đến thiếu nghiêm trọng hồng cầu khỏe mạnh, cuối cùng gây ra những biểu hiện nặng.
Những thay đổi di truyền trên gây sai lệch quá trình phát triển của tế bào hồng cầu. Tế bào hồng cầu chưa trưởng thành của người bệnh gọi là nguyên bào hồng cầu có hình dạng bất thường hoặc có nhân thừa. Những nguyên bào hồng cầu bất thường này không thể phát triển thành tế bào hồng cầu trưởng thành và mất chức năng. Thiếu tế bào hồng cầu khỏe mạnh dẫn đến các triệu chứng đặc trưng của bệnh thiếu máu.
Chẩn đoán
Bệnh có thể được chẩn đoán lâm sàng bởi các triệu chứng đặc trưng như vàng da, gan và lách to, dị tật xương ngón tay, ngón chân. Tuy nhiên các triệu chứng trên dễ nhầm lẫn với các bệnh khác. Một số xét nghiệm bổ sung bao gồm:
- Xét nghiệm công thức máu toàn phần, kiểm tra tủy xương
- Xét nghiệm bilirubin huyết thanh
- Đo lượng sắt trong cơ thể
- Xét nghiệm di truyền tìm đột biến gen gây bệnh
Người bệnh cần cung cấp thêm thông tin về tiền sử bệnh của gia đình nhằm hỗ trợ quá trình chẩn đoán.
Điều trị
Hiện nay, cấy ghép tế bào gốc tạo máu là phương pháp điều trị hoàn toàn bệnh thiếu máu rối loạn sinh hồng cầu bẩm sinh. Trường hợp bệnh nhẹ hơn có thể áp dụng một số phương pháp sau:
- Truyền máu và lọc máu
- Thuốc, ví dụ như chất thải sắt hoặc interferon alpha-2A (chỉ điều trị bệnh loại I)
- Phẫu thuật cắt bỏ lá lách hoặc túi mật
Người bệnh cần khám sức khỏe định kỳ, phòng ngừa biến chứng. Các vấn đề về gan, nội tiết tố và tim ngày càng trở nên nghiêm trọng theo thời gian, do lượng hemoglobin thấp và hàm lượng sắt cao.
Dạng di truyền
Thiếu máu rối loạn sinh hồng cầu bẩm sinh có nhiều kiểu di truyền khác nhau.
- Bệnh loại 1 và 2 di truyền theo kiểu lặn trên nhiễm sắc thể thường. Do đó, bệnh chỉ biểu hiện khi có cả hai bản sao của gen đột biến trong mỗi tế bào. Bệnh nhân mắc bệnh lặn trên nhiễm sắc thể thường sẽ có bố và mẹ mang một bản sao của gen đột biến, nhưng bố mẹ ít khi biểu hiện triệu chứng bệnh.
- Một số trường hợp, bệnh loại 3 di truyền theo kiểu trội trên nhiễm sắc thể thường, chỉ cần một bản sao của gen đột biến trong mỗi tế bào đủ để gây bệnh. Người bị bệnh thường có cha mẹ và người thân mắc bệnh.
- Dựa vào các ca bệnh được ghi nhận, người ta cho rằng bệnh loại 4 do đột biến mới (denove) trong gen KLF1 và xảy ra ở những người không có tiền sử mắc chứng rối loạn này trong gia đình họ.

Nguồn: U.S. National Library of Medicine

Nguồn: U.S. National Library of Medicine
Phòng ngừa
Bệnh thiếu máu rối loạn sinh hồng cầu bẩm sinh có dạng di truyền phức tạp. Các thành viên trong gia đình nên khám sức khỏe và tầm soát bệnh định kỳ nếu có thành viên mắc bệnh. Nếu cha hoặc mẹ mắc bệnh, con sinh ra sẽ có 50% khả năng di truyền bệnh. Do đó, để bảo đảm 100% khả năng con không bị bệnh, cha mẹ có thể chọn phương pháp thụ tinh nhân tạo IVF và sàng lọc phôi PGS/PGD.
Trường hợp bệnh di truyền lặn đột biến gen FGT1A1, cha mẹ mang đột biến dị hợp nên gần như không có biểu hiện bệnh, do đó rất khó phát hiện cho đến khi sinh con. Để chủ động phòng ngừa, cha mẹ nên làm xét nghiệm sàng lọc gen lặn để chủ động cho tương lai của con.
Các cặp vợ chồng trước khi mang thai cần tư vấn và xét nghiệm di truyền đảm bảo sinh con khỏe mạnh.
Các tên gọi khác
- Anemia, dyserythropoietic, congenital
- CDA
References
- Genetic Testing Information. Congenital dyserythropoietic anemia. Retrieved September 23, 2022 from https://www.ncbi.nlm.nih.gov/gtr/conditions/C0002876/
- Genetic and Rare Diseases Information Center. Congenital dyserythropoietic anemia. Retrieved September 23, 2022 from https://rarediseases.info.nih.gov/diseases/1999/congenital-dyserythropoietic-anemia/
- Catalog of Genes and Diseases from OMIM. ANEMIA, CONGENITAL DYSERYTHROPOIETIC, TYPE Ia. Retrieved September 23, 2022 from https://omim.org/entry/224120
- Catalog of Genes and Diseases from OMIM. ANEMIA, CONGENITAL DYSERYTHROPOIETIC, TYPE II. Retrieved September 23, 2022 from https://omim.org/entry/224100
- Catalog of Genes and Diseases from OMIM. ANEMIA, CONGENITAL DYSERYTHROPOIETIC, TYPE III. Retrieved September 23, 2022 from https://omim.org/entry/105600
- Catalog of Genes and Diseases from OMIM. ANEMIA, CONGENITAL DYSERYTHROPOIETIC, TYPE IV. Retrieved September 26, 2022 from https://www.omim.org/entry/613673
- U.S National Library of Medicine. Congenital dyserythropoietic anemia. Retrieved September 23, 2022 from https://medlineplus.gov/genetics/condition/congenital-dyserythropoietic-anemia/
- National Organization for Rare Disorders. Congenital dyserythropoietic anemia. Retrieved September 23, 2022 from https://rarediseases.org/gard-rare-disease/congenital-dyserythropoietic-anemia/
- Achille Iolascon, Hermann Heimpel, Anders Wahlin and Hannah Tamary. Congenital dyserythropoietic anemias: molecular insights and diagnostic approach. Published September 26, 2013 https://doi.org/10.1182/blood-2013-05-468223
- Orphanet. Nonketotic Hyperglycinemia. Retrieved September 23, 2022 from https://www.orpha.net/consor/cgi-bin/OC_Exp.php?lng=en&Expert=85
- Yaddanapudi Ravindranath, MBBS, Robert M. Johnson, PhD, Gerard Goyette, BS, Steven Buck, MS, Manisha Gadgeel, MD, and Patrick G. Gallagher, MD. KLF1 E325K-associated Congenital Dyserythropoietic Anemia Type IV: Insights Into the Variable Clinical Severity. Published August, 2018 https://doi.org/10.1097/MPH.0000000000001056