Hội chứng Smith-Magenis là một bệnh di truyền ảnh hưởng đến nhiều bộ phận của cơ thể, đặc trưng bởi thiểu năng trí tuệ từ nhẹ đến trung bình, chậm nói và chậm phát triển kĩ năng ngôn ngữ, ngoài ra người bệnh có khuôn mặt dị biệt, bị rối loạn giấc ngủ và những vấn đề hành vi.
Biểu hiện lâm sàng
Hầu hết người mắc hội chứng Smith-Magenis đều có khuôn mặt vuông, rộng cùng với đôi mắt sâu, má đầy đặn và hàm dưới nhô cao, sóng mũi tẹt. Miệng có xu hướng trễ xuống với môi trên đầy đặn và cong hướng ra ngoài. Người bệnh có những khác biệt nhỏ trên khuôn mặt chưa rõ ràng trong giai đoạn sơ sinh nhưng dần trở nên rõ ràng hơn trong giai đoạn đi nhà trẻ và trưởng thành sau này. Họ còn bị bất thường về răng.
Người bệnh sớm có biểu hiện rối loạn giấc ngủ. Họ có thể rất buồn ngủ vào ban ngày nhưng lại khó ngủ vào ban đêm, thức giấc nhiều lần trong đêm cho đến tận trời sáng.
Người mắc hội chứng Smith-Magenis thường có tính cách dễ mến, thu hút nhưng họ cũng có các vấn đề hành vi như nóng nảy và hung hăng, hay lo lắng, bốc đồng và khó tập trung. Họ có thể tự gây thương tích cho chính bản thân như tự cắn, đánh, đập đầu và cào da. Hành vi tự ôm mình lặp đi lặp lại là một đặc điểm có thể chỉ có xuất hiện ở những người bị hội chứng Smith-Magenis. Một số người mắc chứng này cũng bắt buộc phải liếm ngón tay để lật các trang sách và tạp chí (hành vi được gọi là "liếm và lật").
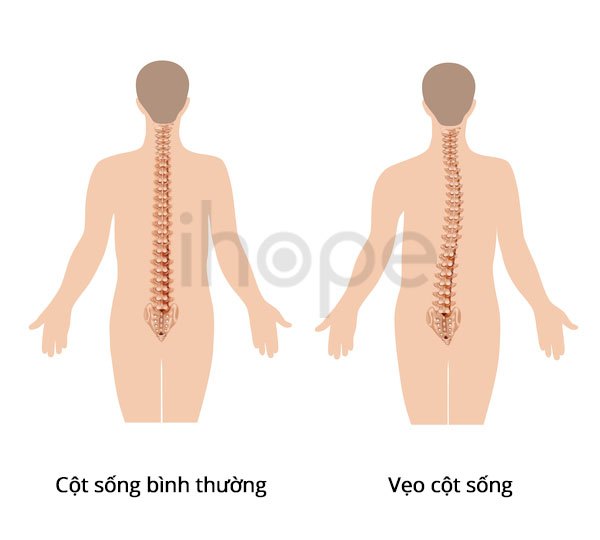
Hội chứng Smith-Magenis có các dấu hiệu và triệu chứng khác như tầm vóc thấp, cột sống cong bất thường (vẹo cột sống), kém nhạy cảm với cơn đau và nhiệt độ, giọng nói khàn. Một số người có bất thường về tai dẫn đến mất thính giác. Người bệnh còn có các bất thường về mắt gây cận thị và các vấn đề thị lực khác. Mặc dù ít phổ biến hơn nhưng cũng có thể xuất hiện các khuyết tật về tim và thận cũng đã được ghi nhận ở những người mắc hội chứng Smith-Magenis.
Nguồn: Alila Medical Media/Shutterstock.com
Độ phổ biến
Hội chứng Smith-Magenis ảnh hưởng đến ít nhất khoảng 1/25.000 người trên toàn thế giới. Tuy nhiên các nhà nghiên cứu tin rằng nhiều người bị hội chứng này nhưng không được chẩn đoán vì vậy nên tỷ lệ mắc thực sự có thể gần khoảng 1/15.000.
Nguyên nhân
Hầu hết những người mắc hội chứng Smith-Magenis do mất một đoạn nhỏ của nhiễm sắc thể số 17 trong mỗi tế bào. Mất đoạn xảy tại vị trí p11.2 trên nhánh ngắn (p) của nhiễm sắc thể 17 . Đoạn bị mất thường có khoảng 3,7 triệu cặp base còn được viết dưới dạng 3,7 megabase (Mb).
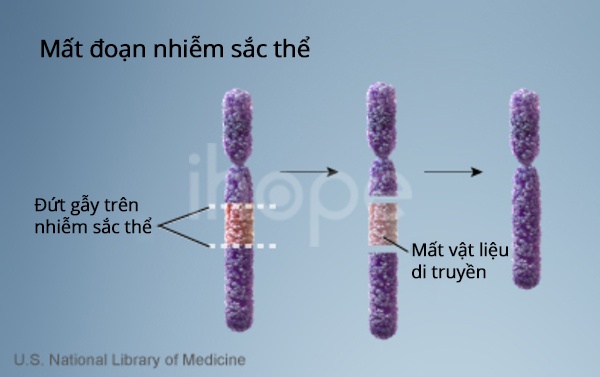
Nguồn: U.S. National Library of Medicine
Một bản sao thừa của đoạn này gây ra dạng bệnh liên quan gọi là hội chứng Potocki-Lupski. Đôi khi đoạn bị mất lớn hơn hoặc nhỏ hơn. Mất đoạn đều tác động đến một trong hai bản sao của nhiễm sắc thể 17 trong mỗi tế bào.
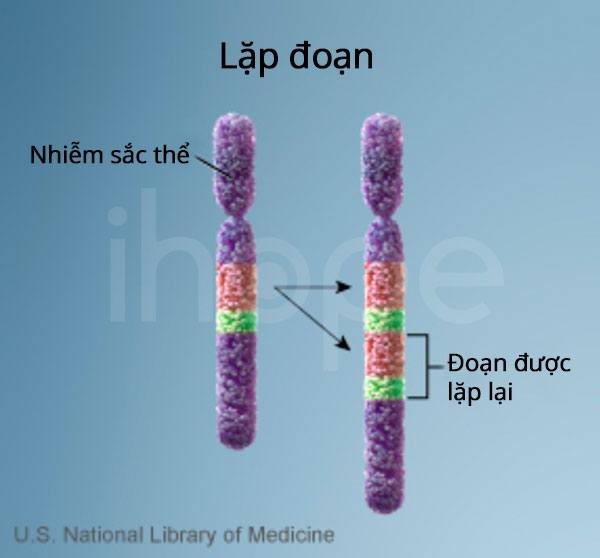
Nguồn: U.S. National Library of Medicine
Mặc dù đoạn bị mất chứa nhiều gen, nhưng người ta tin rằng mất gen RAI1 là nguyên nhân chính gây ra nhiều biểu hiện đặc trưng của hội chứng Smith-Magenis. Tất cả các trường hợp mất đoạn gây ra hội chứng đều chứa gen này. Gen RAI1 mang thông tin tổng hợp ra một loại protein giúp điều hòa hoạt động của các gen khác. Mặc dù hầu hết các gen được điều hòa bởi protein RAI1 chưa được xác định, nhưng protein này dường như còn kiểm soát hoạt động của một số gen liên quan đến nhịp sinh học hàng ngày như chu kỳ ngủ thức. Nghiên cứu cho thấy mất gen RAI1 dẫn đến giảm lượng protein RAI1 trong tế bào và gián đoạn biểu hiện của các gen ảnh hưởng đến nhịp sinh học. Những thay đổi này có thể giải thích cho tình trạng rối loạn giấc ngủ xảy ra với hội chứng Smith-Magenis. Hiện nay người ta vẫn chưa rõ làm thế nào mà mất một bản sao của gen RAI1 lại dẫn đến các vấn đề khác về thể chất, tinh thần và hành vi.
Một tỷ lệ nhỏ những người mắc hội chứng Smith-Magenis có gen RAI1 bị đột biến thay vì mất đoạn nhiễm sắc thể chứa gen này. Mặc dù họ có nhiều đặc điểm chính của hội chứng nhưng họ có nguy cơ thấp biểu hiện các đặc điểm như tầm vóc thấp, mất tính giác và bất thường tim hoặc thận so với những người bị mất gen này. Có khả năng mất đoạn khiến họ bị xóa thêm các gen khác, từ đó phá sinh thêm các dấu hiệu và triệu chứng. Hiện nay, vai trò của những gen này vẫn đang được nghiên cứu.
Dạng di truyền
Hội chứng Smith-Magenis thường không di truyền. Nguyên nhân thường do mất đoạn nhiễm sắc thể hoặc đột biến gen RAI1 xảy ra trong quá trình hình thành các tế bào sinh sản (trứng hoặc tinh trùng) hoặc trong giai đoạn phát triển sớm của bào thai. Hầu hết những người mắc hội chứng Smith-Magenis không có tiền sử bệnh trong gia đình.
Trong một số ít trường hợp, người mắc hội chứng Smith-Magenis đã thừa hưởng mất đoạn hoặc đột biến từ người mẹ không bị bệnh vì người mẹ này chỉ có biến đổi di truyền trong tế bào trứng. Hiện tượng này được gọi là khảm dòng mầm.
Phòng ngừa
Nếu bạn lo lắng về tiền sử gia đình mắc hội chứng Smith-Magenis hoặc nếu bạn đã có con mắc chứng rối loạn này, hãy cân nhắc nói chuyện với bác sĩ hoặc chuyên gia tư vấn di truyền để được giúp lập kế hoạch mang thai trong tương lai.
Hiện nay, xét nghiệm sàng lọc không xâm lấn NIPT ihope có thể phát hiện hội chứng Smith-Magenis do mất vi đoạn, thực hiện sớm từ tuần thai thứ 10 với độ chính xác lên đến 99%. Chỉ cần lấy 5ml máu thai phụ để xét nghiệm nên bảo đảm an toàn cho cả mẹ và con.
Các tên gọi khác
- Hội chứng mất đoạn 17p-
- Monosomy 17p11.2
- Hội chứng mất đoạn nhiễm sắc thể 17p
- Monosomy một phần 17p
- SMS
References
- Genetic Testing Information. Smith-Magenis syndrome. Retrieved Nov 2, 2020 from https://www.ncbi.nlm.nih.gov/gtr/conditions/C0795864/
- Catalog of Genes and Diseases from OMIM. SMITH-MAGENIS SYNDROME; SMS. Retrieved Nov 2, 2020 from https://omim.org/entry/182290
- Genetic and Rare Diseases Information Center. Smith-Magenis syndrome. Retrieved Nov 2, 2020 from https://rarediseases.info.nih.gov/diseases/8197/smith-magenis-syndrome
- U.S National Library of Medicine. Smith-Magenis syndrome. Retrieved Nov 2, 2020 from https://medlineplus.gov/genetics/condition/smith-magenis-syndrome/
- National Organization for Rare Disorders. Smith-Magenis syndrome. Retrieved Nov 2, 2020 from https://rarediseases.org/rare-diseases/smith-magenis-syndrome/
- Smith-Magenis Syndrome (SMS) Foundation UK. What is SMS? Retrieved Nov 2, 2020 from https://smith-magenis.org/what-is-sms/
Related posts
-

Hội chứng DiGeorge (mất đoạn 22q11.2)
Bất thường cấu trúc NST -
Hội chứng Angelman
Bất thường cấu trúc NST -
Hội chứng Down
Bất thường số lượng NST -
Hội chứng Cri-du-chat
Bất thường cấu trúc NST -
Thay đổi số lượng nhiễm sắc thể ảnh hưởng thế nào?
Đột biến và bệnh -
Alpha thalassemia – Thiếu máu tán huyết
Đột biến lặn