
Hội chứng Angelman là một bệnh di truyền phức tạp ảnh hưởng chủ yếu đến hệ thần kinh. Hội chứng này có các đặc điểm đặc trưng như chậm phát triển, thiểu năng trí tuệ, khiếm khuyết nói rất nặng và các vấn đề về cử động và thăng bằng. Hầu hết trẻ em bị hội chứng này cũng bị co giật tái phát (động kinh) và đầu nhỏ (tật đầu nhỏ) . Chậm phát triển rõ rệt ở độ tuổi từ 6 đến 12 tháng, các dấu hiệu và triệu chứng phổ biến khác thường xuất hiện ở thời thơ ấu.
Ảnh: Tật đầu nhỏ
Nguồn: Elements of Morphology, National Human Genome Research Institute

Ảnh: Da và tóc sáng màu
Nguồn: Natalia_R/Shutterstock.com
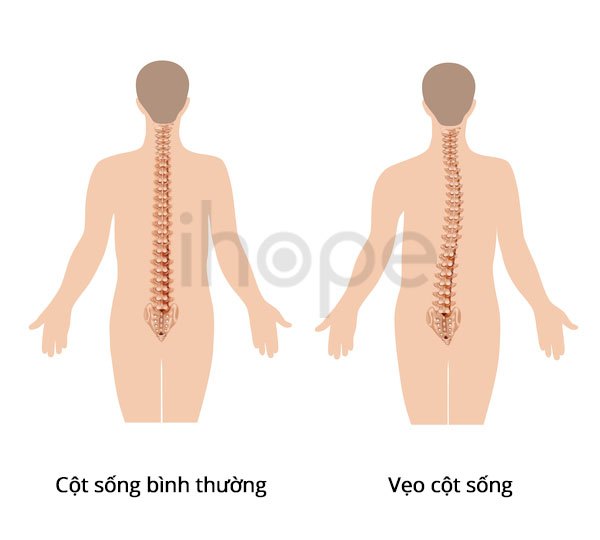
Ảnh: Chứng vẹo cột sống
Nguồn: Alila Medical Media/Shutterstock.com
Biểu hiện lâm sàng
Trẻ em mắc hội chứng Angelman thường có trạng thái hay vui vẻ, dễ bị kích động, hay cười và vỗ tay thường xuyên. Tăng động, hay mất tập trung và thích nước là các triệu chứng phổ biến. Hầu hết trẻ em bị bệnh cũng khó ngủ và thời gian cần ngủ ít hơn bình thường.
Theo tuổi tác, những người mắc hội chứng Angelman trở nên ít bị kích động và giấc ngủ được cải thiện hơn. Tuy nhiên, những người bị bệnh vẫn tiếp tục bị thiểu năng trí tuệ, khiếm thính trầm trọng và co giật trong suốt cuộc đời của họ. Người lớn mắc hội chứng Angelman có các đặc điểm đặc biêt trên khuôn mặt có thể được mô tả là "thô". Các đặc điểm phổ biến khác như làn da trắng bất thường với mái tóc sáng màu và cột sống cong bất thường từ bên này sang bên kia (chứng vẹo cột sống) . Nhìn chung, tuổi thọ của họ gần như bình thường.
Triệu chứng
Các dấu hiệu và triệu chứng của hội chứng Angelman bao gồm:
- Động kinh, thường bắt đầu từ 2 đến 3 tuổi
- Chuyển động cứng hoặc giật
- Kích thước đầu nhỏ, có độ phẳng ở phía sau đầu
- Đẩy lưỡi
- Tóc, da và mắt có màu sáng
- Các hành vi bất thường, chẳng hạn như vỗ tay và nâng cao cánh tay khi đi bộ
- Các vấn đề về giấc ngủ
Các biến chứng
Các biến chứng liên quan đến hội chứng Angelman bao gồm:
- Khó cho ăn. Khó khăn trong việc phối hợp giữa việc bú và nuốt có thể gây ra các vấn đề về bú ở trẻ sơ sinh. Bác sĩ nhi khoa có thể đề nghị một loại sữa công thức có hàm lượng calo cao để giúp bé tăng cân.
- Tăng động. Trẻ mắc hội chứng Angelman thường di chuyển nhanh chóng từ hoạt động này sang hoạt động khác, khoảng thời gian chú ý ngắn và ngậm tay hoặc đồ chơi trong miệng. Tăng động thường giảm theo độ tuổi và thường không cần dùng thuốc.
- Rối loạn giấc ngủ. Những người mắc hội chứng Angelman thường có kiểu ngủ-thức bất thường và có thể ngủ ít hơn hầu hết mọi người. Khó ngủ có thể cải thiện theo tuổi tác. Thuốc và liệu pháp hành vi có thể giúp kiểm soát rối loạn giấc ngủ.
- Cột sống cong (vẹo cột sống). Một số người mắc hội chứng Angelman phát triển cột sống bất thường cong từ bên này sang bên kia theo thời gian.
- Béo phì. Trẻ lớn hơn mắc hội chứng Angelman có xu hướng thèm ăn lớn, có thể dẫn đến béo phì.
Độ phổ biến
Ước tính khoảng 1/12.000 đến 1/20.000 số người bị mắc hội chứng này trên toàn cầu.
Nguyên nhân
Nhiều triệu chứng đặc trưng của hội chứng Angelman do một gen gọi là UBE3A bị mất chức năng. Mỗi người thừa hưởng một bản sao của gen UBE3A từ cha và mẹ, cả hai bản sao của đều được kích hoạt trong nhiều mô của cơ thể. Tuy nhiên, ở một số vùng nhất định của não, chỉ bản sao được thừa hưởng từ mẹ mới hoạt động. Nếu bản sao gen UBE3A của mẹ bị mất do thay đổi nhiễm sắc thể hoặc đột biến gen, người con sẽ không có bản sao hoạt động của gen trong một số phần của não.
Một số cơ chế di truyền khác nhau có thể bất hoạt hoặc gây mất bản sao gen UBE3A từ mẹ. Hầu hết các trường hợp hội chứng Angelman (khoảng 70%) xảy ra khi một đoạn nhiễm sắc thể 15 của người mẹ có chứa gen này bị xóa. Trong các trường hợp khác (khoảng 11%), hội chứng Angelman là do đột biến trong bản sao gen UBE3A của mẹ.
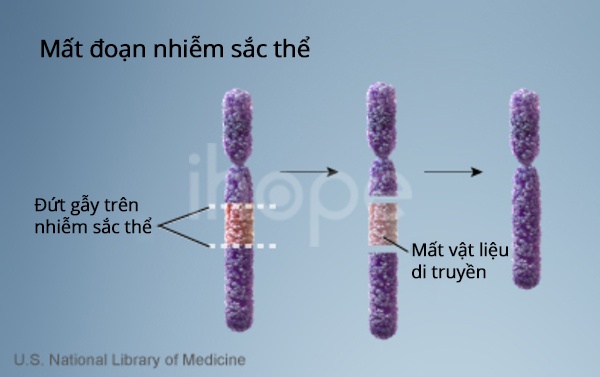
Nguồn: Credit: U.S. National Library of Medicine
Một tỷ lệ nhỏ trong các trường hợp bị bệnh, hội chứng Angelman xuất hiện do một người thừa hưởng hai bản sao nhiễm sắc thể số 15 từ cha mình (bản sao của người cha) thay vì một bản sao từ cha hoặc mẹ. Hiện tượng này được gọi là hiện tượng di truyền đơn thai ở cha. Hiếm khi, hội chứng Angelman cũng có thể được gây ra bởi sự sắp xếp lại nhiễm sắc thể được gọi là chuyển vị, hoặc do đột biến hoặc khiếm khuyết khác trong vùng ADN kiểm soát sự hoạt hóa của gen UBE3A. Những thay đổi di truyền này có thể làm tắt (bất hoạt) UBE3A hoặc các gen khác trên bản sao nhiễm sắc thể 15 của mẹ một cách bất thường.
Các nguyên nhân của hội chứng Angelman chưa được tìm ra trong số từ 10-15% số người bị ảnh hưởng. Những thay đổi liên quan đến các gen hoặc nhiễm sắc thể khác có thể là nguyên nhân gây ra rối loạn trong những trường hợp này.
Ở một số người mắc hội chứng Angelman, việc mất một gen gọi là OCA2 có liên quan đến tóc sáng màu và làn da trắng. Gen OCA2 nằm trên đoạn của nhiễm sắc thể số 15 thường bị mất ở những người mắc chứng rối loạn này. Tuy nhiên, mất gen OCA2 không gây ra các dấu hiệu và triệu chứng khác của hội chứng Angelman. Protein được tạo ra từ gen này giúp xác định màu sắc (sắc tố) của da, tóc và mắt.
Chẩn đoán
Bác sĩ có thể nghi ngờ hội chứng Angelman nếu trẻ bị chậm phát triển và các dấu hiệu và triệu chứng khác của bệnh như khó cử động và giữ thăng bằng, kích thước đầu nhỏ, phẳng ở phía sau đầu và hay cười.
Xét nghiệm di truyền có thể xác định những bất thường trong nhiễm sắc thể, dấu hiệu của hội chứng Angelman.
Kết hợp của các xét nghiệm di truyền có thể cho biết các khiếm khuyết nhiễm sắc thể liên quan đến hội chứng Angelman. Các xét nghiệm có thể xem xét thực hiện như:
- Xét nghiệm ADN cha mẹ, sàng lọc ba trong bốn bất thường di truyền đã biết gây ra hội chứng Angelman
- Xét nghiệm bất thường nhiễm sắc thể
- Xét nghiệm đột biến gen. Hiếm khi hội chứng Angelman xảy ra khi bản sao gen UBE3A của một người đang hoạt động nhưng bị đột biến. Nếu kết quả xét nghiệm ADN bình thường, bác sĩ có thể yêu cầu xét nghiệm giải trình tự gen UBE3A để tìm đột biến ở mẹ.
Điều trị
Hiện tại, bệnh chưa thể chữa trị khỏi. Các nghiên cứu tập trung vào xác định các gen cụ thể để điều trị. Phương pháp điều trị hiện nay tập trung vào dùng thuốc và kiểm soát các vấn đề liên quan đến sự phát triển liên quan đến bệnh.
Tùy thuộc vào các dấu hiệu và triệu chứng của trẻ, phương án điều trị có thể bao gồm:
- Thuốc chống co giật
- Liệu pháp vật lý cải thiện các vấn đề liên quan đến di chuyển và vận động
- Liệu pháp giao tiếp có thể bao gồm giao tiếp bằng hình ảnh và kí hiệu
- Liệu pháp hành vi giúp vượt qua chứng tăng động, kém tập trung và hỗ trợ phát triển
Dạng di truyền
Hầu hết các trường hợp hội chứng Angelman không phải do di truyền, đặc biệt là những trường hợp do mất đoạn nhiễm sắc thể số 15 của người mẹ hoặc do mất đoạn nhiễm sắc thể đơn tính của cha. Những thay đổi di truyền xảy ra như những sự kiện ngẫu nhiên trong quá trình hình thành các tế bào sinh sản (trứng và tinh trùng) hoặc trong quá trình phát triển phôi sớm. Những người bị ảnh hưởng thường không có tiền sử rối loạn trong gia đình của họ.
Nguồn: U.S. National Library of Medicine
Hiếm khi, một đột biến di truyền gây ra hội chứng Angelman có thể được di truyền. Ví dụ, có thể xảy ra đột biến trên gen UBE3A hoặc trong vùng lân cận của ADN kiểm soát sự hoạt hóa gen được truyền từ thế hệ này sang thế hệ khác.
Phòng ngừa
Trong một số trường hợp hiếm hoi, hội chứng Angelman có thể di truyền từ cha mẹ bị ảnh hưởng sang con cái thông qua các gen khiếm khuyết. Nếu bạn lo lắng về tiền sử gia đình mắc hội chứng Angelman hoặc nếu bạn đã có con mắc chứng rối loạn này, hãy cân nhắc nói chuyện với bác sĩ hoặc chuyên gia tư vấn di truyền để được giúp lập kế hoạch mang thai trong tương lai.
Hiện nay, xét nghiệm sàng lọc không xâm lấn NIPT ihope có thể phát hiện hội chứng Angelman do mất vi đoạn, thực hiện sớm từ tuần thai thứ 10 với độ chính xác lên đến 99%. Chỉ cần lấy 5ml máu thai phụ để xét nghiệm nên bảo đảm an toàn cho cả mẹ và con.
Các tên gọi khác
- AS
References
- Genetic Testing Information. Angelman syndrome. Retrieved July 14, 2020 from https://www.ncbi.nlm.nih.gov/gtr/conditions/C0162635/
- Catalog of Genes and Diseases from OMIM. ANGELMAN SYNDROME; AS. Retrieved July 14, 2020 from https://omim.org/entry/105830
- Genetic and Rare Diseases Information Center. Angelman syndrome. Retrieved Retrieved July 14, 2020 from https://rarediseases.info.nih.gov/diseases/5810/angelman-syndrome
- U.S. National Library of Medicine. Angelman Syndrome. Retrieved July 14, 2020 from https://medlineplus.gov/genetics/condition/angelman-syndrome/
- Families. Research. Clinic. Community. Angelman. Retrieved July 14, 2020 from https://www.angelman.org/
- The Canadian Angleman Syndrome Society. Keeping families strong. Retrieved July 14, 2020 from https://www.angelmancanada.org/
- Foundation for Angelman Syndrome Therapeutics. Angel Syndrome. Retrieved July 14, 2020 from https://cureangelman.org/
- Medical Home Portal.Angel Syndrome. Retrieved July 14, 2020 from https://www.medicalhomeportal.org/diagnoses-and-conditions/angelman-syndrome
- National Organization for Rare Disorders. Angleman Syndrome. Retrieved July 14, 2020 from https://www.cdc.gov/hepatitis/hcv/index.htm





