Hội chứng DiGeorge hay hội chứng mất đoạn 22q11.2 là một bệnh di truyền do mất đoạn nhỏ của nhiễm sắc thể 22. Đoạn bị mất nằm gần tâm nhiễm sắc thể tại vị trí q11.2. Các triệu chứng của hội chứng DiGeorge rất đa dạng, chúng có thể ảnh hưởng đến phần lớn mọi bộ phận của cơ thể.
Nguồn: U.S. National Library of Medicine
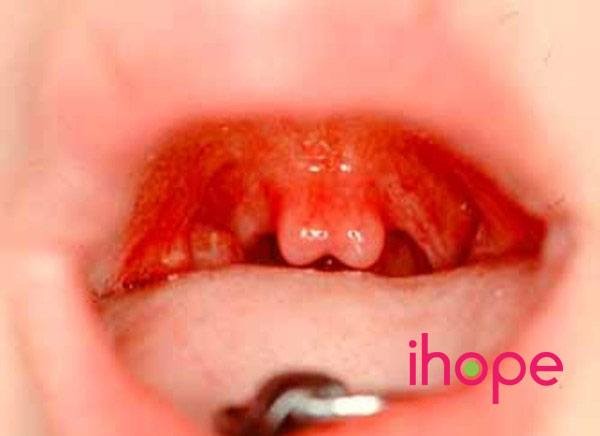
Ảnh: Uvula hai bên
Nguồn: Elements of Morphology, National Human Genome Research Institute
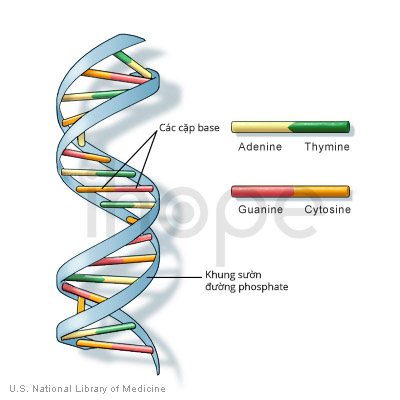
Ảnh: Cấu trúc ADN
Nguồn: U.S. National Library of Medicine
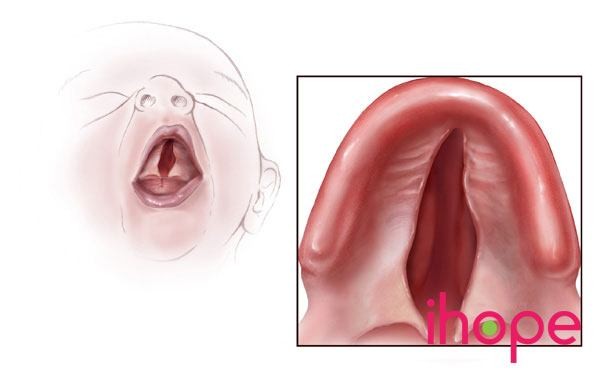
Ảnh: Hở hàm ếch
Nguồn: Centers for Disease Control and Prevention, National Center on Birth Defects and Developmental Disabilities
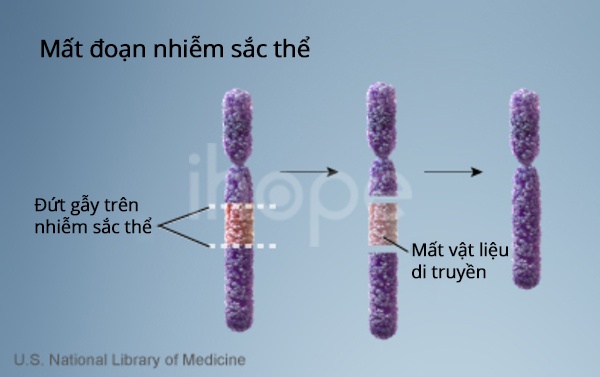
Ảnh: Mất vi đoạn
Nguồn: U.S. National Library of Medicine
Biểu hiện lâm sàng
Nngười mắc hội chứng mất đoạn 22q11.2 thường có các biểu hiện bao gồm bất thường tim thường từ khi mới sinh, nhiễm trùng tái phát do các vấn đề với hệ miễn dịch và khuôn mặt dị biệt. Với người bệnh, các cơ tạo thành vòm miệng (vòm miệng) có thể không khép lại hoàn toàn mặc dù các mô bao phủ chúng, do đó họ bị hở hàm ếch dưới niêm mạc. Vòm miệng bất thường thường cong cao và có thể có một vết nứt tại phần mô mềm treo phía sau miệng (uvula hai bên) . Sứt môi dưới niêm mạc cũng có thể cản trở khả năng nói, bởi vì không khí thoát ra khỏi mũi khi phát âm, dẫn đến giọng mũi. Người bệnh cũng có thể gặp các vấn đề hô hấp, bất thường thận, nồng độ canxi trong máu thấp (có thể dẫn đến co giật), giảm tiểu cầu, ăn uống rất khó khăn, vấn đề tiêu hóa và mất thính giác. Ngoài ra, vấn đề với xương có thể gây ra biểu hiện tầm vóc thấp bé nhẹ cân và bất thường cột sống.
Nhiều trẻ em mắc hội chứng mất đoạn 22q11.2 bị chậm phát triển bao gồm chậm nói, chậm tăng trưởng và phát triển, một số trẻ bị thiểu năng trí tuệ nhẹ hoặc khó học tập. Khi lớn tuổi hơn, chúng gặp khó khăn khi đọc, làm bài tập toán học và giải quyết vấn đề. Trẻ thường cần được giúp đỡ để thay đổi và điều chỉnh hành vi của mình khi phản ứng với các tình huống. Ngoài ra, trẻ có nguy cơ cao bị suy giảm trí nhớ, tăng động (ADHD), giảm chú ý và tự kỷ.
Bởi vì các dấu hiệu và triệu chứng của hội chứng mất đoạn 22q11.2 rất đa dạng, các nhóm đặc điểm khác nhau từng được mô tả như những bệnh lý riêng biệt như hội chứng DiGeorge, hội chứng cơ tim (còn gọi là hội chứng Shprintzen) và hội chứng khuôn mặt dị thường kết mạc. Ngoài ra, một số trẻ mất đoạn 22q11.2 được chẩn đoán mắc hội chứng Opitz G/BBB và hội chứng tim Cayler. Khi nguyên nhân di truyền được làm sáng tỏ, các bệnh lý trên được xem như một hội chứng duy nhất với nhiều dấu hiệu và triệu chứng có thể xảy ra. Để tránh nhầm lẫn, bệnh thường được gọi là hội chứng mất đoạn 22q11.2 dựa trên nguyên nhân di truyền cơ bản của nó.
Các triệu chứng
Các dấu hiệu và triệu chứng của hội chứng DiGeorge (hội chứng mất đoạn 22q11.2) có các biểu hiện và mức độ nghiêm trọng khác nhau, tùy thuộc vào thể trạng của từng người và mức độ tác động nghiêm trọng của các khiếm khuyết. Một số dấu hiệu và triệu chứng có thể rõ ràng khi mới sinh, nhưng những dấu hiệu khác có thể không biểu hiện cho đến khi trẻ lớn hơn hoặc trong giai đoạn thời thơ ấu.
Các dấu hiệu và triệu chứng có thể xuất hiện như sau:
- Tiếng tim đập và da hơi xanh do máu giàu oxy lưu thông kém (tím tái) do dị tật tim
- Nhiễm trùng thường xuyên
- Một số đặc điểm trên khuôn mặt, chẳng hạn như cằm kém phát triển, tai thấp, mắt mở to hoặc rãnh hẹp tại môi trên
- Một khoảng trống trên vòm miệng (hở hàm ếch) hoặc các vấn đề khác với vòm miệng
- Tăng trưởng chậm
- Ăn uống khó khăn, chậm tăng cân hoặc các vấn đề tiêu hóa
- Các vấn đề hô hấp
- Trương lực cơ kém
- Chậm phát triển, chẳng hạn như chậm vận động, ngồi dậy chậm hoặc chậm trong các giai đoạn phát triển quan trọng khác của trẻ sơ sinh
- Chậm phát triển giọng nói hoặc giọng mũi, nói ngọng
- Chậm tiếp thu hoặc thiểu năng
- Các vấn đề hành vi
Biến chứng
Các phần của nhiễm sắc thể số 22 bị mất trong hội chứng DiGeorge (hội chứng mất đoạn 22q11.2) tham gia trong quá trình phát triển của một số quy trình của cơ thể. Do đó, mất đoạn 22q11.2 có thể gây ra một số sai sót trong quá trình phát triển của thai nhi.
Các biểu hiện phổ biến bao gồm:
- Dị tật tim. Hội chứng mất đoạn 22q11.2 thường gây ra dị tật tim nên tim không thể cung cấp đủ lượng máu giàu oxy. Ví dụ, các khuyết tật có thể bao gồm một lỗ giữa các ngăn dưới của tim (thông liên thất); chỉ có một mạch lớn, chứ không phải hai mạch dẫn ra khỏi tim (ống động mạch thân); hoặc sự kết hợp của bốn cấu trúc bất thường của tim (tứ chứng Fallot).
- Suy tuyến cận giáp. Bốn tuyến cận giáp tại cổ có chức năng điều chỉnh hàm lượng canxi và photpho trong cơ thể. Hội chứng mất đoạn 22q11.2 khiến các tuyến cận giáp nhỏ hơn bình thường nên lượng hormone tuyến cận giáp (PTH) suy giảm nghiêm trọng, do đó người bệnh bị suy tuyến cận giáp. Họ có lượng canxi thấp và lượng photpho trong máu cao.
- Rối loạn chức năng tuyến ức. Tuyến ức nằm bên dưới xương ức là nơi sản xuất các tế bào T—một loại tế bào bạch cầu trưởng thành. Tế bào T trưởng thành giúp cơ thể chống lại nhiễm trùng. Với trẻ em mắc hội chứng mất đoạn 22q11.2, tuyến ức có thể nhỏ hoặc thiếu nên chúng có chức năng miễn dịch kém và thường xuyên bị nhiễm trùng nặng.
- Sứt môi. Một biểu hiện phổ biến của hội chứng mất đoạn 22q11.2 là hở hàm ếch—một khe hở tại vòm miệng, có hoặc không có khe hở môi. Những bất thường vòm miệng khác ít thấy hơn có thể khiến người bệnh khó nuốt hoặc tạo ra một số âm thanh lạ khi nói.
- Khuôn mặt dị biệt. Người bệnh có một số đặc điểm bao gồm tai nhỏ và thấp, độ rộng của lỗ mắt ngắn (khe hở vòm miệng), mắt bị che, khuôn mặt tương đối dài, đầu mũi nở rộng (hình củ) hoặc rãnh ngắn hoặc dẹt tại môi trên.
- Vấn đề học tập, hành vi và sức khỏe tâm thần. Mất đoạn 22q11.2 ảnh hưởng quá trình phát triển và hoạt động của não, nên người bệnh gặp các vấn đề với học tập, giao tiếp xã hội, phát triển hoặc hành vi. Biểu hiện phổ biến gồm chậm nói và khó học tập. Một số trẻ phát triển chứng rối loạn tăng động giảm chú ý (ADHD) hoặc tự kỷ. Càng về sau, nguy cơ trầm cảm, rối loạn lo âu và các rối loạn sức khỏe tâm thần khác càng tăng.
- Rối loạn tự miễn. Người bệnh có chức năng miễn dịch kém khi còn nhỏ do tuyến ức nhỏ hoặc thiếu, nên họ có nguy cơ cao mắc bệnh tự miễn như viêm khớp dạng thấp hoặc bệnh Graves.
- Các vấn đề khác. Nhiều biểu hiện khác có thể liên quan đến hội chứng mất đoạn 22q11.2, chẳng hạn như khiếm thính, thị lực kém, vấn đề hô hấp, chức năng thận kém và tầm vóc tương đối thấp.
Độ phổ biến
Ước tính hội chứng mất đoạn 22q11.2 ảnh hưởng khoảng 1/4.000 trên toàn dân số. Tuy nhiên, số ca bệnh thực tế có thể nhiều hơn ước tính này bởi vì nhiều trường hợp có thể bị bỏ qua do các biểu hiện đa dạng của bệnh. Ví dụ như những người có dấu hiệu và triệu chứng nhẹ, hoặc bệnh có thể bị nhầm lẫn với các bệnh khác có biểu hiện tương tự.
Nguyên nhân
Phần lớn những người mắc hội chứng mất đoạn 22q11.2 đều thiếu một chuỗi gồm khoảng 3 triệu cặp codon ADN (các cặp base ) trên một bản sao của nhiễm sắc thể 22 trong mỗi tế bào. Vùng này chứa từ 30 đến 40 gen, nhiều gen chưa được xác định rõ ràng.
Các nhà nghiên cứu đang cố gắng để xác định tất cả các gen góp phần vào các đặc điểm của hội chứng mất đoạn 22q11.2. Họ nhận thấy rằng mất một gen trên nhiễm sắc thể 22, TBX1 có lẽ là nguyên nhân gây ra nhiều dấu hiệu đặc trưng của hội chứng (chẳng hạn như dị tật tim, hở hàm ếch , khuôn mặt dị biệt, khiếm thính và mức canxi thấp). Một số nghiên cứu cho thấy mất gen này cũng có thể góp phần gây ra các vấn đề hành vi. Mất gen COMT trong cùng vùng của nhiễm sắc thể 22 cũng có thể giải thích nguy cơ gia tăng các vấn đề hành vi và bệnh tâm thần. Mất thêm các gen trong vùng bị mất đoạn có thể góp phần vào các đặc điểm khác nhau của hội chứng mất đoạn 22q11.2.
Chẩn đoán
Xét nghiệm di truyền để xác nhận mất đoạn 22q11.2 thường được thực hiện từ một mẫu máu được gửi đến phòng thí nghiệm lâm sàng. Thử nghiệm được đề xuất ngày nay bao gồm các phương pháp phức tạp hơn phương pháp lai huỳnh quang tại chỗ (FISH) được sử dụng trước đây vì các thử nghiệm mới cũng có thể cho biết quy mô của việc xóa hoặc sao chép. Chúng bao gồm các phương pháp “toàn bộ bộ gen” chẳng hạn như phép lai gen so sánh (CGH), microarray SNP (ví dụ: CGA) hoặc xét nghiệm MLPA. Một lần nữa, các nghiên cứu FISH được nhắm mục tiêu có thể bỏ sót một số thao tác xóa/sao chép 22q11.2 “lồng nhau” nhỏ hơn mà các phương pháp phức tạp hơn sẽ phát hiện. Phương pháp karyotype chỉ rất hiếm khi có thể phát hiện mất đoạn 22q11.2 trong khoảng 25% trường hợp và do đó không phải là phương pháp được khuyến nghị để phát hiện 22q11.2DS.
Điều trị
Cũng giống như mỗi đứa trẻ bình thường đều khác nhau, mỗi đứa trẻ bị mất đoạn 22q11.2 sẽ có những nhu cầu phát triển và y tế khác nhau. Với suy nghĩ này, trẻ em bị mất đoạn cần được chăm sóc bởi các chuyên gia thích hợp, nhưng ngoài ra, việc được đội ngũ y tế khám để được chăm sóc phối hợp thường rất hữu ích. Mặc dù không có “cách chữa trị” hội chứng mất đoạn 22q11.2, có nhiều cách để quản lý các vấn đề mà nó gây ra và các triệu chứng được phát hiện càng sớm càng tốt.
Sự can thiệp có thể được thực hiện theo một trong hai cách: liệu pháp tư nhân, có hoặc không có bảo hiểm, hoặc các dịch vụ can thiệp sớm thông qua tiểu bang hoặc quận. Sự can thiệp sớm có thể được yêu cầu bởi bác sĩ nhi khoa hoặc nhà cung cấp dịch vụ chăm sóc sức khỏe khác hoặc trực tiếp bởi cha mẹ. Việc đánh giá bao gồm phương pháp tiếp cận nhóm với các chuyên gia liên quan đến trị liệu ngôn ngữ và âm ngữ, vật lý trị liệu (PT), trị liệu nghề nghiệp (OT), giáo dục đặc biệt,...
Dạng di truyền
Hội chứng mất đoạn 22q11.2 là đột biến di truyền trội trên nhiễm sắc thể thường mất đoạn trên một bản sao của nhiễm sắc thể số 22 trong mỗi tế bào đủ để gây ra tình trạng này. Tuy nhiên, phần lớn các trường hợp bị mất đoạn 22q11.2 không được di truyền. Mất đoạn thường xảy ra một cách ngẫu nhiên trong quá trình hình thành các tế bào sinh sản (trứng hoặc tinh trùng) hoặc trong giai đoạn phát triển sớm của bào thai. Người bệnh thường không có tiền sử bệnh trong gia đình của họ, mặc dù họ có thể truyền bệnh cho con. Trong khoảng 10% các trường hợp, người bệnh thừa hưởng nhiễm sắc thể 22 bị mất đoạn từ cha mẹ. Trong trường hợp di truyền, các thành viên khác trong gia đình cũng có thể bị ảnh hưởng.
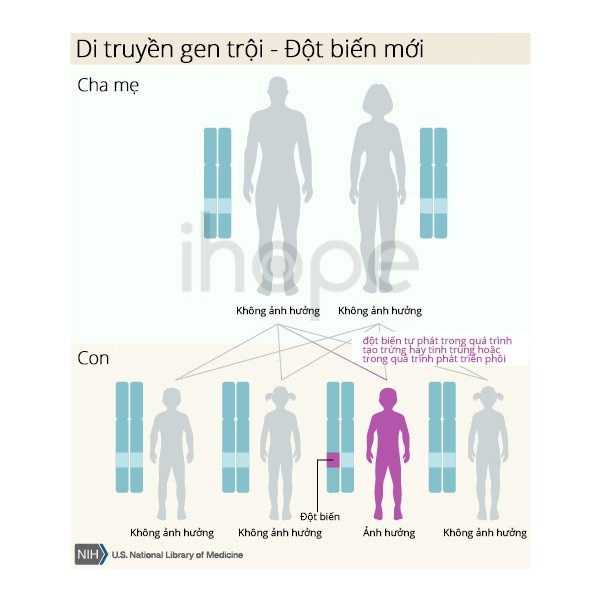
Nguồn: U.S. National Library of Medicine
Phòng ngừa
Trong một số trường hợp, hội chứng DiGeorge (hội chứng mất đoạn 22q11.2) có thể được truyền từ cha mẹ sang con cái. Nếu lo lắng về tiền sử gia đình mắc hội chứng mất đoạn 22q11.2 hoặc nếu đã có con mắc hội chứng này, cha mẹ nên tham vấn ý kiến từ bác sĩ chuyên về bệnh di truyền (nhà di truyền học) hoặc chuyên gia tư vấn di truyền để được trợ giúp cho những lần mang thai trong tương lai.
Hiện nay, xét nghiệm sàng lọc không xâm lấn NIPT ihope có thể phát hiện hội chứng DiGeorge do mất vi đoạn, thực hiện sớm từ tuần thai thứ 10 với độ chính xác lên đến 99%. Chỉ cần lấy 5ml máu thai phụ để xét nghiệm nên bảo đảm an toàn cho cả mẹ và con.
Các tên gọi khác
- 22q11.2DS
- Hội chứng Opitz G/BBB chiếm ưu thế trên NST thường
- CATCH22
- Hội chứng tim Cayler
- Hội chứng khuôn mặt dị thường conotruncal (CTAF)
- Hội chứng mất đoạn 22q11.2
- Hội chứng DiGeorge
- Hội chứng Sedlackova
- Hội chứng Shprintzen
- VCFS
- Hội chứng velo-tim-mặt
- Hội chứng cơ tim
References
- NCBI. 22q11.2 Deletion Syndrome . Retrieved July 14, 2020 from https://www.ncbi.nlm.nih.gov/books/NBK1523
- OMIM. DiGeorge syndrome; DGS. Retrieved July 14, 2020 from http://omim.org/entry/188400
- National Organization For Rare Disorders. Chromosome 22q11.2 Deletion Syndrome. Retrieved July 14, 2020 from https://rarediseases.info.nih.gov/diseases/10299/index
- U.S. National Library of Medicine. 22q11.2 deletion syndrome. Retrieved July 14, 2020 from https://medlineplus.gov/genetics/condition/22q112-deletion-syndrome/
- The Virtual Center For VCFS. Exciting News for People with VCFS and Mental Illness. Retrieved July 14, 2020 from https://www.vcfscenter.com/
- International 22q11.2 Foundation. Diagnosis of 22q11.2 Deletion. Retrieved June 24, 2024 from https://22q.org/what-is-22q/diagnosis/
- International 22q11.2 Foundation. Medical Treatment. Retrieved June 24, 2024 from https://22q.org/symptoms-care/medical-treatment/