Loạn dưỡng cơ Duchenne là một nhóm bệnh di truyền gây ra tình trạng yếu cơ và teo cơ. Bệnh ảnh hưởng chủ yếu đến cơ xương (cơ sử dụng để vận động) và cơ tim. Dạng loạn dưỡng cơ này hầu như chỉ xảy ra ở nam giới.
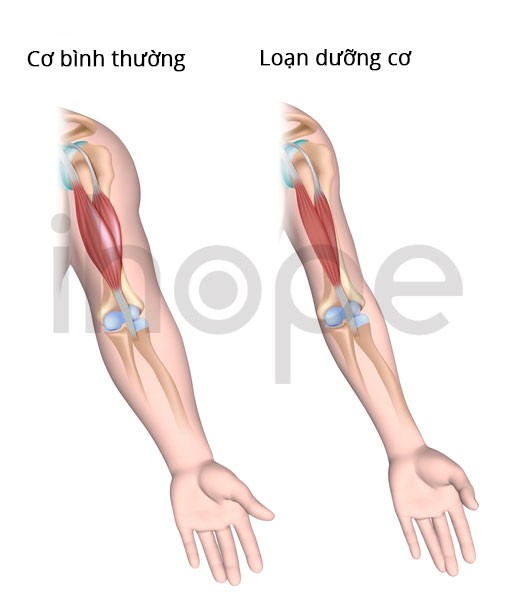
Nguồn: U.S. National Library of Medicine.
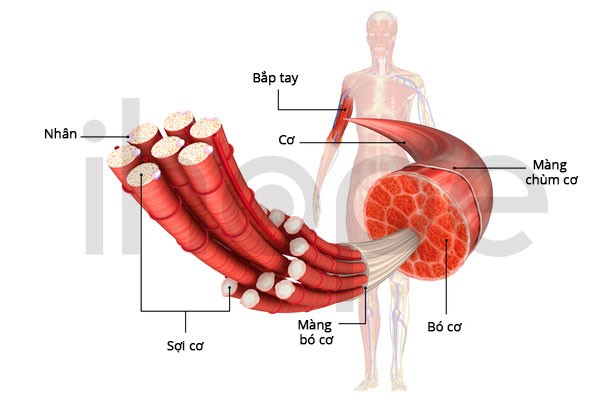
Ảnh: Cấu trúc cơ xương.
Nguồn: U.S. National Library of Medicine.
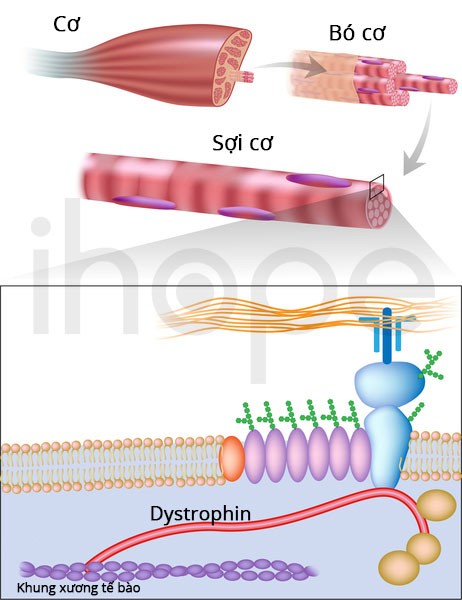
Ảnh: Dystrophin trong sợi cơ.
Nguồn: U.S. National Library of Medicine.
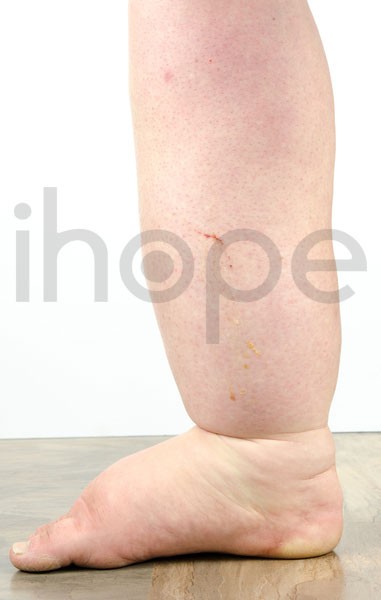
Ảnh: Phù chân và bàn chân.
Nguồn: U.S. National Library of Medicine.
Biểu hiện lâm sàng
Các dấu hiệu và triệu chứng của bệnh loạn dưỡng cơ Duchenne do các đột biến gen gây ra. Ở các bé trai mắc chứng loạn dưỡng cơ Duchenne, tình trạng yếu cơ có xu hướng xuất hiện trong thời thơ ấu và trở nên trầm trọng một cách nhanh chóng. Trẻ em mắc bệnh có thể bị chậm các kỹ năng vận động, chẳng hạn như ngồi, đứng và đi. Trẻ thường phụ thuộc vào xe lăn ở tuổi vị thành niên.
Loạn dưỡng cơ Duchenne thường liên quan đến bệnh cơ tim. Dạng bệnh tim này làm suy yếu cơ tim, ảnh hưởng đến chức năng bơm máu của tim. Ở chứng loạn dưỡng cơ Duchenne, bệnh cơ tim thường bắt đầu ở tuổi thiếu niên. Sau đó, cơ tim phình to ra, và phát triển thành bệnh cơ tim giãn. Các dấu hiệu và triệu chứng của bệnh cơ tim giãn bao gồm nhịp tim không đều (loạn nhịp tim), khó thở, cực kỳ mệt mỏi, phù chân và bàn chân . Những vấn đề về tim có thể trở nên xấu đi và nguy hiểm đến tính mạng. Nam giới mắc chứng loạn dưỡng cơ Duchenne thường sống đến 20 tuổi.
Một tình trạng liên quan được gọi là bệnh cơ tim giãn liên kết X là một dạng bệnh tim gây ra bởi các đột biến trong cùng một gen như loạn dưỡng cơ Duchenne, và đôi khi nó được phân loại là loạn dưỡng cơ Becker cận lâm sàng. Những người bị bệnh cơ tim giãn nở liên kết X thường không bị teo cơ, mặc dù họ có thể có những thay đổi nhỏ trong tế bào cơ xương có thể phát hiện được thông qua xét nghiệm trong phòng thí nghiệm.
Độ phổ biến
Bệnh loạn dưỡng cơ Duchenne ảnh hưởng đến 1 / 3.500-5.000 bé trai trên toàn thế giới. Mỗi năm, Hoa Kỳ có khoảng 400 đến 600 bé trai sinh ra bị mắc căn bệnh này.
Nguyên nhân
Các đột biến trên gen DMD gây ra bệnh loạn dưỡng cơ Duchenne. Gen DMD cung cấp hướng dẫn để tạo ra một loại protein gọi là dystrophin . Protein này nằm chủ yếu ở cơ xương và cơ tim, nơi nó giúp ổn định và bảo vệ các sợi cơ. Dystrophin cũng có thể đóng một vai trò trong việc truyền tín hiệu hóa học trong tế bào.
Các đột biến trên gen DMD làm thay đổi cấu trúc và chức năng của dystrophin hoặc ngăn chặn việc sản xuất dystrophin. Các tế bào cơ không có đủ lượng protein này sẽ bị tổn thương khi cơ hoạt động liên tục. Theo thời gian, các sợi cơ bị tổn thương, dẫn đến yếu cơ và gây ra các vấn đề về tim, triệu chứng của chứng loạn dưỡng cơ Duchenne.
Vì chứng loạn dưỡng cơ Duchenne là kết quả của việc tạo ra dystrophin bị lỗi, những tình trạng này được phân loại là chứng loạn dưỡng cơ.
Chẩn đoán
Khi chẩn đoán bất kỳ dạng loạn dưỡng cơ nào, các phát hiện lâm sàng, tiền sử gia đình, nồng độ creatine kinase trong máu, yếu tố di truyền và sinh thiết cơ thường được sử dụng để chẩn đoán bệnh.
Tiền sử bệnh
Trước khi thực hiện các xét nghiệm chuyên sâu, bác sĩ thường hỏi về tình trạng sức khỏe, các triệu chứng mà bệnh nhân gặp phải, tiền sử bệnh của bệnh nhân và gia đình và thực hiện khám sức khỏe bao gồm chứng giả phì đại cơ, lệch cột sống thắt lưng, bất thường về dáng đi và mức độ phản xạ cơ.
Nồng độ creatine kinase trong máu
Đầu quá trình chẩn đoán, bác sĩ thường yêu cầu xét nghiệm máu để kiểm tra nồng độ creatine kinase. Creatine kinase là một loại enzyme được giải phóng khi cơ bị tổn thương. Khi nồng độ creatine kinase trong máu tăng cao, có nghĩa là cơ đang bị phân hủy bởi một số bất thường trong cơ thể, như chứng loạn dưỡng cơ hoặc viêm. Mức độ creatine kinase cao có thể được phát hiện trước khi xuất hiện các triệu chứng.
Mức creatine kinase thường đạt đỉnh ở khi trẻ 2 tuổi, sau đó giảm dần với tốc độ 25% mỗi năm, cuối cùng trở lại mức bình thường khi một lượng mô cơ đã được thay thế bằng mô sẹo.
Xét nghiệm di truyền
Xét nghiệm di truyền liên quan đến việc phân tích ADN của bất kỳ tế bào nào (thường là tế bào máu) để xem liệu có đột biến trên gen DMD hay không. Thông thường, xét nghiệm di truyền được chỉ định cho những bệnh nhân có nồng độ creatine kinas trong máu cao và có biểu hiện lâm sàng về bệnh loạn dưỡng cơ. Một người mang đột biến trên gen DMD có thể được xác nhận mắc bệnh loạn dưỡng cơ Duchenne.
Sinh thiết cơ
Để có thêm thông tin, bác sĩ có thể yêu cầu sinh thiết cơ. Một mẫu cơ được lấy để tìm các bất thường của dystrophin trong cơ. Protein dystrophin có thể được quan sát bằng cách nhuộm mẫu cơ với một loại thuốc nhuộm đặc biệt. Một người mắc bệnh sẽ không có dystrophin và không có lớp đệm xung quanh các tế bào cơ.
Các kỹ thuật hiện đại có thể sử dụng sinh thiết để phân biệt chứng loạn dưỡng cơ với các bệnh do viêm và các bệnh khác, đồng thời để phân biệt giữa các dạng loạn dưỡng cơ khác nhau.
Nếu nghi ngờ bệnh nhân mắc bệnh loạn dưỡng cơ Duchenne mặc dù kết quả phân tích di truyền âm tính, việc phát hiện dystrophin bằng kỹ thuật Western blot hoặc nhuộm với kháng thể chọn lọc được thực hiện trong mô lấy từ sinh thiết cơ.
Điều trị
Các phương pháp điều trị loạn dưỡng cơ Dunchenne là nhằm mục đích cải thiện các triệu chứng của bệnh.
Điều trị bênh cơ tim giãn bằng cách sử dụng thuốc chống sung huyết, có thể ghép tim trong trường hợp bệnh nặng.
Sử dụng các thiết bị trợ giúp cho các biến chứng hô hấp, đặt biệt là ban đêm.
Sử dụng một số loại thuốc như:
- Thuốc prednisone - một loại steroid - được dùng để cải thiện chức năng ở những người mắc bệnh. Prednisone đã được chứng minh là kéo dài khả năng đi lại từ 2 đến 5 năm. Tuy nhiên, các tác dụng phụ có thể có của prednisone bao gồm tăng cân, huyết áp cao, thay đổi hành vi và chậm phát triển.
- Deflazacort- một dạng tổng hợp của prednisilone, được sử dụng ở châu Âu và được cho là có ít tác dụng phụ hơn so với prednisone.
- Cyclosporin - được sử dụng và đã cải thiện chức năng lâm sàng ở trẻ em, nhưng việc sử dụng nó còn gây tranh cãi.
- Oxandrolone - một loại thuốc được sử dụng trong một nghiên cứu, có tác dụng tương tự như prednisone với ít tác dụng phụ hơn. Một số liệu pháp khác cũng đang được nghiên cứu, bao gồm coenzyme Q10, glutamine, pentoxifylline và PTC124.
Vật lý trị liệu được sử dụng để thúc đẩy khả năng vận động và ngăn ngừa chứng co cứng.
Phẫu thuật có thể được thực hiện đối với những trường hợp co cứng và vẹo cột sống nghiêm trọng.
Dạng di truyền
Loạn dưỡng cơ Duchenne được di truyền theo kiểu gen lặn liên kết X. Gen liên quan đến tình trạng này nằm trên nhiễm sắc thể X, một trong hai nhiễm sắc thể giới tính. Ở nam giới (chỉ có một nhiễm sắc thể X), một bản sao bị đột biến trong mỗi tế bào là đủ để gây ra căn bệnh này. Ở phụ nữ (những người có hai nhiễm sắc thể X), bệnh xảy ra khi đột biến xảy ra ở cả hai bản sao của gen. Do đó, bệnh thường mắc phải ở nam giới nhiều hơn so với nữ giới. Một đặc điểm của di truyền liên kết X là người cha không thể truyền tính trạng liên kết X cho con trai của họ.
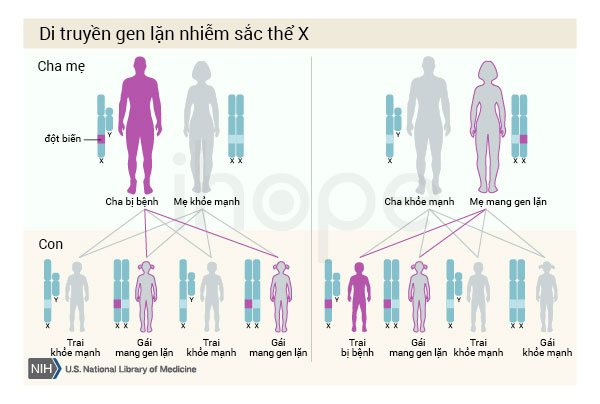
Nguồn: U.S. National Library of Medicine
Trong nhiều trường hợp, một người đàn ông mắc bệnh do thừa hưởng đột biến từ mẹ. Phần còn lại của các trường hợp có thể là do đột biến gen mới và không được di truyền.
Trong di truyền lặn liên kết X, nữ giới có một bản sao của gen bị đột biến trong mỗi tế bào được gọi là người mang gen. Người này có thể di truyền gen bị đột biến nhưng thường không có các dấu hiệu và triệu chứng của bệnh. Tuy nhiên, đôi khi phụ nữ mang đột biến gen DMD có thể bị yếu cơ và chuột rút. Những triệu chứng này thường nhẹ hơn so với những tình trạng ở nam giới. Phụ nữ mang đột biến gen DMD cũng có nguy cơ phát triển các bất thường về tim.
Phòng ngừa
Bệnh có cơ chế di truyền liên kết X phức tạp nên khó phát hiện ở những người phụ nữ chỉ mang một đột biến gen lặn, đến khi có con mới biết được thì đã quá muộn. Do đó, các cặp vợ chồng trước khi mang thai cần làm sàng lọc gen lặn để sàng lọc bệnh loạn dưỡng cơ Duchenne, đảm bảo sinh con khỏe mạnh và lành lặn.
References
- Genetic Testing Information. Duchenne muscular dystrophy. Retrieved April 27, 2021 from https://www.ncbi.nlm.nih.gov/gtr/conditions/C0013264/
- Genetic Testing Information. Becker muscular dystrophy. Retrieved April 27, 2021 from https://www.ncbi.nlm.nih.gov/gtr/conditions/C0917713/
- Catalog of Genes and Diseases from OMIM. MUSCULAR DYSTROPHY, DUCHENNE TYPE; DMD . Retrieved April 27, 2021 from https://omim.org/entry/310200
- Catalog of Genes and Diseases from OMIM. EPIDERMOLYSIS BULLOSA, JUNCTIONAL, NON-HERLITZ TYPE. Retrieved April 27, 2021, 2021 from https://omim.org/entry/226650
- U.S. National Library of Medicine. Duchenne and Becker muscular dystrophy. Retrieved April 27, 2021 from https://medlineplus.gov/genetics/condition/duchenne-and-becker-muscular-dystrophy/
- National Human Genome Research Institute. About Duchenne Muscular Dystrophy. Retrieved April 27, 2020 from https://www.genome.gov/Genetic-Disorders/Duchenne-Muscular-Dystrophy
- U.S. National Library of Medicine. Duchenne muscular dystrophy. Retrieved April 27, 2021 from https://medlineplus.gov/ency/article/000705.htm
- Muscular Dystrophy Association. Duchenne Muscular Dystrophy (DMD). Retrieved April 27, 2021 from https://www.mda.org/disease/duchenne-muscular-dystrophy/diagnosis