
Hội chứng Marfan là một bệnh di truyền ảnh hưởng đến các mô liên kết ở nhiều bộ phận của cơ thể. Mô liên kết cung cấp sức mạnh và tính linh hoạt cho các cấu trúc như xương, dây chằng, cơ, mạch máu và van tim. Các dấu hiệu và triệu chứng của hội chứng Marfan rất khác nhau về mức độ nghiêm trọng, thời điểm khởi phát và tốc độ tiến triển.
Biểu hiện lâm sàng
Bởi vì mô liên kết có mặt ở khắp cơ thể, hội chứng Marfan có thể ảnh hưởng đến nhiều cơ quan và bộ phận, như các bất thường về tim, mạch máu, mắt, xương và khớp. Hai đặc điểm chính của hội chứng Marfan là các vấn đề về thị lực do lệch thủy tinh thể (ectopia lentis) ở một hoặc cả hai mắt và khiếm khuyết trong mạch máu lớn phân phối máu từ tim đến phần còn lại của cơ thể (động mạch chủ). Động mạch chủ có thể suy yếu và căng ra, dẫn đến phình thành mạch máu (chứng phình động mạch). Động mạch chủ bị giãn có thể làm hở van động mạch chủ, dẫn đến rách đột ngột các lớp trong thành động mạch chủ (bóc tách động mạch chủ). Phình động mạch chủ và bóc tách có thể đe dọa tính mạng.
Nhiều người bị hội chứng Marfan có thêm các vấn đề về tim bao gồm rò rỉ ở van nối hai trong bốn buồng tim (sa van hai lá) hoặc van điều chỉnh dòng máu từ tim vào động mạch chủ (hở van động mạch chủ). Rò rỉ ở các van này có thể gây ra khó thở, mệt mỏi và nhịp tim không đều hoặc thêm nhịp (đánh trống ngực).
Những người mắc hội chứng Marfan thường cao và mảnh khảnh, có ngón tay và ngón chân thon dài, khớp lỏng lẻo và sải tay vượt quá chiều cao cơ thể. Các đặc điểm chung khác bao gồm khuôn mặt dài và hẹp, răng mọc chen chúc, độ cong bất thường của cột sống (vẹo cột sống), rạn da không liên quan đến tăng hoặc giảm cân và ngực hóp hoặc ngực nhô. Một số bệnh nhân tích tụ không khí bất thường trong khoang ngực có thể dẫn đến xẹp phổi (tràn khí màng phổi tự phát). Một màng được gọi là màng cứng, bao quanh não và tủy sống, có thể mở rộng bất thường (dural ectasia) ở những người mắc hội chứng Marfan. Chứng ectasia có thể gây đau lưng, bụng, chân hoặc đầu. Hầu hết các cá nhân mắc hội chứng Marfan đều bị cận thị. Đục thủy tinh thể có thể xảy ra ở tuổi trưởng thành và tăng áp lực trong mắt (bệnh tăng nhãn áp) xảy ra thường xuyên hơn ở những người mắc hội chứng Marfan so với những người bình thường.
Các đặc điểm của hội chứng Marfan có thể trở nên rõ ràng bất cứ lúc nào từ giai đoạn sơ sinh đến tuổi trưởng thành. Tùy thuộc vào sự khởi phát và mức độ nghiêm trọng của các dấu hiệu và triệu chứng, hội chứng Marfan có thể gây tử vong sớm. Tuy nhiên, với phương pháp điều trị thích hợp, nhiều người bệnh có tuổi thọ bình thường.
Độ phổ biến
Tỉ lệ mắc hội chứng Marfan khoảng 1/5.000 trên toàn thế giới.
Nguyên nhân
Gen FBN1 cung cấp hướng dẫn để tạo ra một loại protein gọi là fibrillin-1. Fibrillin-1 gắn (liên kết) với các protein fibrillin-1 khác và các phân tử khác để tạo thành các sợi nhỏ như sợi chỉ được gọi là microfibrils. Microfibrils trở thành một phần của các sợi cung cấp sức mạnh và tính linh hoạt cho mô liên kết. Ngoài ra, các sợi nhỏ liên kết với các phân tử được gọi là yếu tố tăng trưởng và giải phóng chúng vào những thời điểm khác nhau để kiểm soát sự phát triển và sửa chữa của các mô và cơ quan trên khắp cơ thể. Một đột biến trên gen FBN1 có thể làm giảm số lượng fibrillin-1 chức năng có sẵn để tạo thành các microfibrils, dẫn đến giảm sự hình thành microfibril. Kết quả là microfibrils không thể liên kết với các yếu tố tăng trưởng, do đó các yếu tố tăng trưởng dư thừa và tính đàn hồi ở nhiều mô bị giảm, dẫn đến sự phát triển quá mức và không ổn định của các mô trong hội chứng Marfan.
Chẩn đoán
Lâm sàng
Chẩn đoán lâm sàng dựa trên tiền sử gia đình và các biểu hiện đặc trưng ở mắt, xương và tim mạch, gồm bốn đặc điểm chính:
- Giãn hoặc bóc tách động mạch chủ ở mức độ của xoang Valsava
- Lệch thủy tinh thể (ectopia lentis)
- Đau dạ dày màng cứng được xác định bằng chụp CT hoặc chụp cộng hưởng từ MRI
- 4/8 đặc điểm điển hình của xương
Tiêu chí chính để chẩn đoán cho một thành viên gia đình bao gồm có cha mẹ, con cái hoặc anh chị em có các đặc điểm chính bên trên, sự hiện diện của đột biến FBN-1 được biết là gây ra hội chứng hoặc một dạng haplotype xung quanh FBN-1 được di truyền và xác định ở một bệnh nhân Marfan có gia đình.
Xét nghiệm gen FBN1
Gen FBN1 liên kết chặt chẽ với hội chứng Marfan. Xét nghiệm gen FBN1 xác định được 70-93% các đột biến. Tuy nhiên, những bệnh nhân âm tính với xét nghiệm đột biến gen nên được xem xét để đánh giá các tình trạng khác có các đặc điểm tương tự hội chứng Marfan như hội chứng Dietz, hội chứng Ehlers Danlos và homocystin niệu. Để thiết lập chẩn đoán một cách rõ ràng trong trường hợp không có tiền sử gia đình, cần có biểu hiện chính từ hai cơ quan của cơ thể và sự tham gia của cơ quan thứ ba. Nếu một đột biến gây ra hội chứng Marfan được xác định, chẩn đoán cần một tiêu chí chính và sự tham gia của cơ quan thứ hai.
Để chẩn đoán cho một người thân của bệnh nhân Marfan đòi hỏi sự hiện diện của một tiêu chí chính trong tiền sử gia đình và một tiêu chí chính trong một hệ cơ quan có sự tham gia của cơ quan thứ hai.
Điều trị
Những người mắc hội chứng Marfan được điều trị bởi một nhóm chuyên gia đa ngành bao gồm một nhà di truyền học, bác sĩ tim mạch, bác sĩ nhãn khoa, bác sĩ chỉnh hình và bác sĩ phẫu thuật lồng ngực.
Các vấn đề về mắt thường được điều trị bằng kính. Nếu lệch thủy tinh thể cản trở tầm nhìn hoặc gây ra bệnh tăng nhãn áp, có thể tiến hành phẫu thuật và cấy ghép thủy tinh thể nhân tạo.
Các vấn đề về xương như cong vẹo cột sống và pectus digvatum có thể phải phẫu thuật. Đối với những người có bàn chân phẳng, có thể sử dụng dụng cụ chỉnh hình để giảm mỏi chân và chuột rút.
Thuốc như thuốc chẹn beta, được sử dụng để giảm áp lực lên động mạch chủ tại thời điểm chẩn đoán hoặc khi có sự giãn nở động mạch chủ tiến triển. Phẫu thuật sửa chữa động mạch chủ được thực hiện khi đường kính động mạch chủ lớn hơn 5 cm ở người lớn và thiếu niên, khi đường kính động mạch chủ tăng 1cm mỗi năm hoặc khi có trào ngược động mạch chủ tiến triển.
Giám sát tim mạch bao gồm siêu âm tim hàng năm để theo dõi tình trạng của động mạch chủ. Hiện nay thuốc chẹn beta có thể trì hoãn nhưng không ngăn cản được cuối cùng phải tiến hành phẫu thuật động mạch chủ.
Nghiên cứu gần đây về thuốc chẹn thụ thể Angiotensin II, một loại thuốc huyết áp khác giống như thuốc chẹn beta, đã cho thấy nhiều hứa hẹn bảo vệ động mạch chủ khỏi bị giãn. Các thử nghiệm lâm sàng sẽ sớm được bắt đầu để xem liệu loại thuốc này có thể ngăn ngừa nhu cầu phẫu thuật tốt hơn thuốc chẹn beta hay không.
Những người mắc hội chứng Marfan nên tránh tiếp xúc và các môn thể thao cạnh tranh và tập thể dục đẳng áp như nâng tạ và các hình thức tập thể dục tĩnh khác. Họ có thể tham gia các bài tập aerobic hoặc bơi lội. Họ cũng nên tránh các loại thuốc như thuốc thông mũi và thực phẩm có chứa caffeine có thể dẫn đến tăng huyết áp mãn tính và kéo căng các mô liên kết trong hệ thống tim mạch.
Dạng di truyền
Bệnh được di truyền theo kiểu trội trên nhiễm sắc thể thường, nghĩa là chỉ cần một bản sao của gen bị thay đổi trong mỗi tế bào là đủ để gây ra rối loạn. Khi một người mang đột biến trội sinh con thì mỗi người con có 50% khả năng kế thừa đột biến đó.

Nguồn: U.S. National Library of Medicine
Ít nhất 25% trường hợp hội chứng Marfan là kết quả của một đột biến mới trên gen FBN1. Những trường hợp này xảy ra ở những người không có tiền sử trong gia đình.
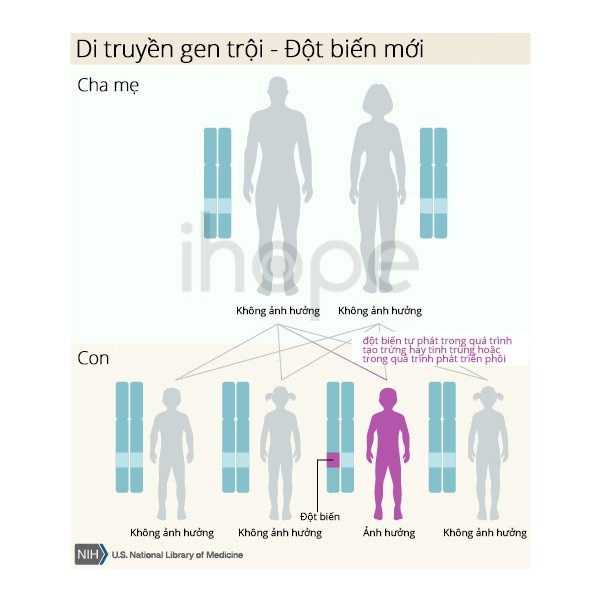
Nguồn: U.S. National Library of Medicine
Phòng ngừa
Nếu một người mang hội chứng Marfan, khi sinh con sẽ có 50% khả năng di truyền bệnh. Do đó, để bảo đảm 100% khả năng con không bị bệnh, cha mẹ có thể chọn phương pháp thụ tinh nhân tạo IVF và sàng lọc phôi PGS/PGD.
Các tên gọi khác
- Marfan's syndrome
- MFS
References
- Genetic Testing Information. Marfan syndrome. Retrieved March 10, 2021 from https://www.ncbi.nlm.nih.gov/gtr/conditions/C0024796/
- Catalog of Genes and Diseases from OMIM. MARFAN SYNDROME; MFS. Retrieved March 10, 2021 from https://omim.org/entry/154700/
- Genetic and Rare Diseases Information Center. Marfan syndrome. Retrieved March 10, 2021 from https://rarediseases.info.nih.gov/diseases/6975/marfan-syndrome/
- U.S. National Library of Medicine. Marfan syndrome. Retrieved March 10, 2021 from https://medlineplus.gov/genetics/condition/marfan-syndrome/
- National Institutes of Health. Marfan syndrome. Retrieved March 10, 2021 from https://www.niams.nih.gov/health-topics/marfan-syndrome/
- Nemours Foundation. Marfan syndrome. Retrieved March 10, 2021 from https://kidshealth.org/en/parents/marfan.html/
- National Human Genome Research Institute. About Marfan Syndrome. Retrieved March 10, 2021 from https://www.genome.gov/Genetic-Disorders/Marfan-Syndrome/
- National Institute of Arthritis and Musculoskeletal and Skin Diseases. What Is Marfan Syndrome? Retrieved March 10, 2021 from https://www.niams.nih.gov/health-topics/marfan-syndrome
- Marfan Foundation. Heart and Blood Vessels in Marfan Syndrome. Retrieved March 10, 2021 from https://www.marfan.org/about/body-systems/heart-and-blood-vessels





