Hội chứng Prader-Willi là một bệnh di truyền phức tạp ảnh hưởng đến nhiều bộ phận của cơ thể. Ở trẻ sơ sinh, bệnh có biểu hiện đặc trưng bởi trương lực cơ yếu (giảm trương lực), bú kém và phát triển chậm. Bắt đầu từ giai đoạn thơ ấu, người bệnh tăng cảm giác thèm ăn vô độ dẫn đến ăn quá nhiều trong thời gian dài và bị béo phì. Một số người mắc hội chứng Prader-Willi đặc biệt là những người bị béo phì cũng khởi phát bệnh tiểu đường loại 2 (dạng bệnh tiểu đường phổ biến nhất).
Biểu hiện lâm sàng
Những người mắc hội chứng Prader-Willi thường bị suy giảm trí tuệ và khuyết tật học tập mức độ từ nhẹ đến trung bình. Các vấn đề về hành vi phổ biến như nóng nảy, bướng bỉnh và hành vi ám ảnh cưỡng chế như gãi vào da. Các bất thường rối loạn giấc ngủ cũng có thể xảy ra. Người bệnh cũng có các đặc trưng trên khuôn mặt như trán hẹp, mắt hình quả hạnh và miệng hình tam giác; tầm vóc thấp bé; và bàn tay và bàn chân nhỏ. Một số người mắc hội chứng Prader-Willi có làn da trắng bất thường và tóc sáng màu. Cả nam và nữ mang bệnh đều có bộ phận sinh dục kém phát triển. Dậy thì chậm hoặc dậy thì không hoàn toàn, và hầu hết những người bị hội chứng này không thể có con hay còn gọi là vô sinh.
Độ phổ biến
Hội chứng Prader-Willi ước tính ảnh hưởng đến khoảng 1/30.000 đến 1/10.000 người trên toàn thế giới.
Nguyên nhân
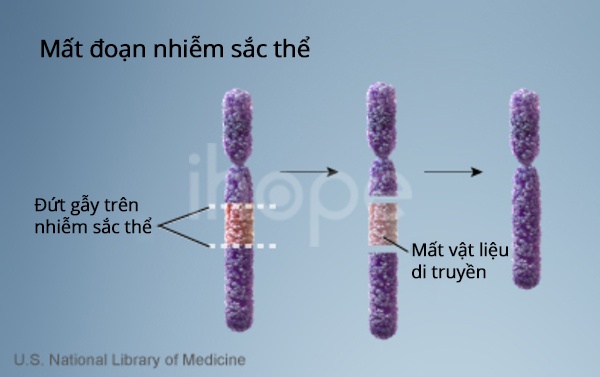
Hội chứng Prader-Willi xảy ra do các gen trong một vùng cụ thể của nhiễm sắc thể số 15 bị mất chức năng. Mỗi người trong chúng ta thường thừa hưởng một bản sao của nhiễm sắc thể này từ cha và mẹ. Một số gen chỉ hoạt động được trên bản sao được thừa hưởng từ cha của một người (hay còn gọi bản sao của người cha). Sự kích hoạt gen cụ thể từ cha mẹ này là do một hiện tượng gọi là in dấu bộ gen.
Hầu hết các trường hợp hội chứng Prader-Willi (chiếm khoảng 70%) xảy ra khi bản sao nhiễm sắc thể số 15 được nhận từ người cha bị mất một đoạn vật liệu di truyền trong mỗi tế bào. Những người có nhiễm sắc thể bị biến đổi này thiếu một số gen quan trọng nhất định trong vùng bị mất vì các gen trên bản sao của cha đã bị mất và các gen trên bản sao của mẹ bị thì không hoạt động). Trong số 25 % trường hợp khác, người mắc hội chứng Prader-Willi có hai bản sao nhiễm sắc thể 15 được thừa hưởng từ mẹ của họ (bản sao của mẹ) thay vì một bản sao từ cha hoặc mẹ. Hiện tượng này được gọi là mẹ không có thai. Rất hiếm nhưng hội chứng Prader-Willi cũng có thể do nhiễm sắc thể sắp xếp lại được gọi là chuyển vị, hoặc do đột biến hoặc khiếm khuyết khác làm tắt (bất hoạt) các gen trên nhiễm sắc thể của người cha một cách bất thường. Hội chứng này do mất chức năng của một số gen trên nhiễm sắc thể số 15.
Nguồn: U.S. National Library of Medicine
Có vẻ như các đặc điểm đặc trưng của hội chứng Prader-Willi là do một số gen trên nhiễm sắc thể 15 bị mất chức năng. Trong số những gen này có những gen mang thông tin tổng hợp ra các phân tử được gọi là ARN nucleolar nhỏ (snoARN). Các phân tử này có nhiều chức năng khác nhau bao gồm giúp điều chỉnh các loại phân tử ARN khác (các phân tử RNA đóng vai trò thiết yếu trong việc sản xuất protein và trong các hoạt động khác của tế bào.).
Các nghiên cứu cho thấy mất một nhóm gen snoARN gọi là cụm SNORD116, có thể đóng vai trò chính trong việc gây ra các dấu hiệu và triệu chứng của hội chứng Prader-Willi. Tuy nhiên, vẫn chưa biết bằng cách nào mà một cụm SNORD116 bị thiếu có thể góp phần gây ra khuyết tật trí tuệ, các vấn đề về hành vi và các đặc điểm thể chất khác của bệnh. Ở một số người mắc hội chứng Prader-Willi, mất gen OCA2 có liên quan đến làn da trắng bất thường và tóc sáng màu. Gen OCA2 nằm trên một đoạn thường bị mất của nhiễm sắc thể số 15 ở những người mắc chứng rối loạn này. Tuy nhiên, mất gen OCA2 không gây ra các dấu hiệu và triệu chứng khác của hội chứng Prader-Willi. Protein được tạo ra từ gen này giúp xác định màu sắc (sắc tố) của da, tóc và mắt.

Nguồn: Natalia_R/Shutterstock.com
Các nhà nghiên cứu đang nghiên cứu các gen khác trên nhiễm sắc thể số 15 cũng có thể liên quan đến các dấu hiệu và triệu chứng chính của bệnh.
Chẩn đoán
Trong nhiều trường hợp hội chứng Prader-Willi, chẩn đoán bệnh dựa trên các biểu hiện lâm sàng ở trẻ sơ sinh.
Nếu trẻ không thể bú trong vài ngày, cơ thể "mềm", yếu ớt, bác sĩ có thể tiến hành xét nghiệm di truyền hội chứng Prader-Willi. Chẩn đoán chính thức phụ thuộc vào độ tuổi của từng bệnh nhân, khi được 3 tuổi. Trước 3 tuổi, triệu chứng quan trọng nhất là trương lực cơ cực kỳ kém, được gọi là giảm trương lực cơ, khiến trẻ sơ sinh cảm thấy mềm nhũn. Ở trẻ từ 3 tuổi trở lên, các triệu chứng khác trở nên rõ ràng, chẳng hạn như béo phì, chậm phát triển trí tuệ, khuyết tật học tập hoặc các vấn đề về hành vi, đặc biệt là liên quan đến thực phẩm và ăn uống.
Điều trị
Hiện nay chưa có cách chữa hội chứng Prader-Willi, mà chủ yếu tập trung vào chữa trị các triệu chứng ở mỗi trường hợp cụ thể.
- Sử dụng núm vú hoặc ống đặc biệt cho những trường hợp khó bú
- Giám sát chặt chẽ lượng thức ăn hàng ngày
- Chế độ ăn uống cân bằng, ít calo và tập thể dục thường xuyên
- Liệu pháp hormone tăng trưởng
- Điều trị các vấn đề về mắt với bác sĩ nhãn khoa
- Điều trị cong cột sống với bác sĩ chấn thương chỉnh hình
- Nghiên cứu giấc ngủ và điều trị
- Vật lý trị liệu
- Liệu pháp hành vi
- Thuốc ức chế tái hấp thu serotonin
- Các chương trình can thiệp sớm hoặc các nhu cầu đặc biệt
- Giáo dục đặc biệt
- Điều trị hormone sinh dục hoặc phẫu thuật điều chỉnh
- Thay thế các hormone sinh dục
Dạng di truyền
Hầu hết các trường hợp hội chứng Prader-Willi không di truyền, đặc biệt là những trường hợp gây ra do mất đoạn ở nhiễm sắc thể số 15 của người cha hoặc do sự suy giảm của người mẹ. Những thay đổi di truyền này xảy ra như những sự kiện ngẫu nhiên trong quá trình hình thành các tế bào sinh sản (trứng và tinh trùng) hoặc trong quá trình phát triển phôi sớm. Những người bị ảnh hưởng thường không có tiền sử rối loạn này trong gia đình của họ.
Nguồn: U.S. National Library of Medicine
Hiếm khi một sự thay đổi gen gây ra hội chứng Prader-Willi có thể được di truyền. Ví dụ, có thể xảy ra một sự thay đổi gen làm bất hoạt các gen trên nhiễm sắc thể số 15 của người cha được truyền từ thế hệ này sang thế hệ khác.
Phòng ngừa
Trong một số trường hợp hiếm hoi, hội chứng Prader-Willi có thể di truyền từ cha mẹ bị ảnh hưởng sang con cái thông qua các gen khiếm khuyết. Nếu bạn lo lắng về tiền sử gia đình mắc hội chứng Prader-Willi hoặc nếu bạn đã có con mắc chứng rối loạn này, hãy cân nhắc nói chuyện với bác sĩ hoặc chuyên gia tư vấn di truyền để được giúp lập kế hoạch mang thai trong tương lai.
Hiện nay, xét nghiệm sàng lọc không xâm lấn NIPT ihope có thể phát hiện hội chứng Prader-Willi do mất vi đoạn, thực hiện sớm từ tuần thai thứ 10 với độ chính xác lên đến 99%. Chỉ cần lấy 5ml máu thai phụ để xét nghiệm nên bảo đảm an toàn cho cả mẹ và con.
Các tên gọi khác
- Hội chứng Prader-Labhart-Willi
- PWS
- Hội chứng Willi-Prader
References
- Genetic Testing Information. Prader-Willi syndrome. Retrieved October 29, 2020 from https://www.ncbi.nlm.nih.gov/gtr/conditions/C0032897/
- Catalog of Genes and Diseases from OMIM. PRADER-WILLI SYNDROME; PWS. Retrieved March 2, 2021 from https://omim.org/entry/176270
- Genetic and Rare Diseases Information Center. Prader-Willi syndrome. Retrieved October 29, 2020 from https://rarediseases.info.nih.gov/diseases/5575/prader-willi-syndrome
- U.S National Library of Medicine. Migraine. Retrieved April 7, 2021 from https://medlineplus.gov/praderwillisyndrome.html
- U.S National Library of Medicine. Prader-Willi syndrome. Retrieved October 29, 2020 from https://medlineplus.gov/genetics/condition/prader-willi-syndrome/
- Mayo Clinic. Prader-Willi syndrome. Retrieved October 29, 2020 from https://www.mayoclinic.org/diseases-conditions/prader-willi-syndrome/symptoms-causes/syc-20355997
- National Center for Biotechnology Information. Prader-Willi syndrome. Retrieved October 29, 2020 from https://www.ncbi.nlm.nih.gov/books/NBK22195/
- Eunice Kennedy Shriver National Institute of Child Health and Human Development. What Are the Treatments for Prader-Willi Syndrome (PWS)? Retrieved October 29, 2020 from https://www.nichd.nih.gov/health/topics/prader-willi/conditioninfo/treatments
- Prader-Willi Syndrome Association. What is Prader-Willi Syndrome? Retrieved October 29, 2020 from https://www.pwsausa.org/what-is-prader-willi-syndrome/
Related posts
-

Hội chứng DiGeorge (mất đoạn 22q11.2)
Bất thường cấu trúc NST -
Hội chứng Angelman
Bất thường cấu trúc NST -
Hội chứng Cri-du-chat
Bất thường cấu trúc NST -
Hội chứng Turner
Bất thường số lượng NST -
Thay đổi cấu trúc nhiễm sắc thể ảnh hưởng thế nào?
Đột biến và bệnh -
Vì sao phải làm xét nghiệm sàng lọc thai?
Sàng lọc NIPT