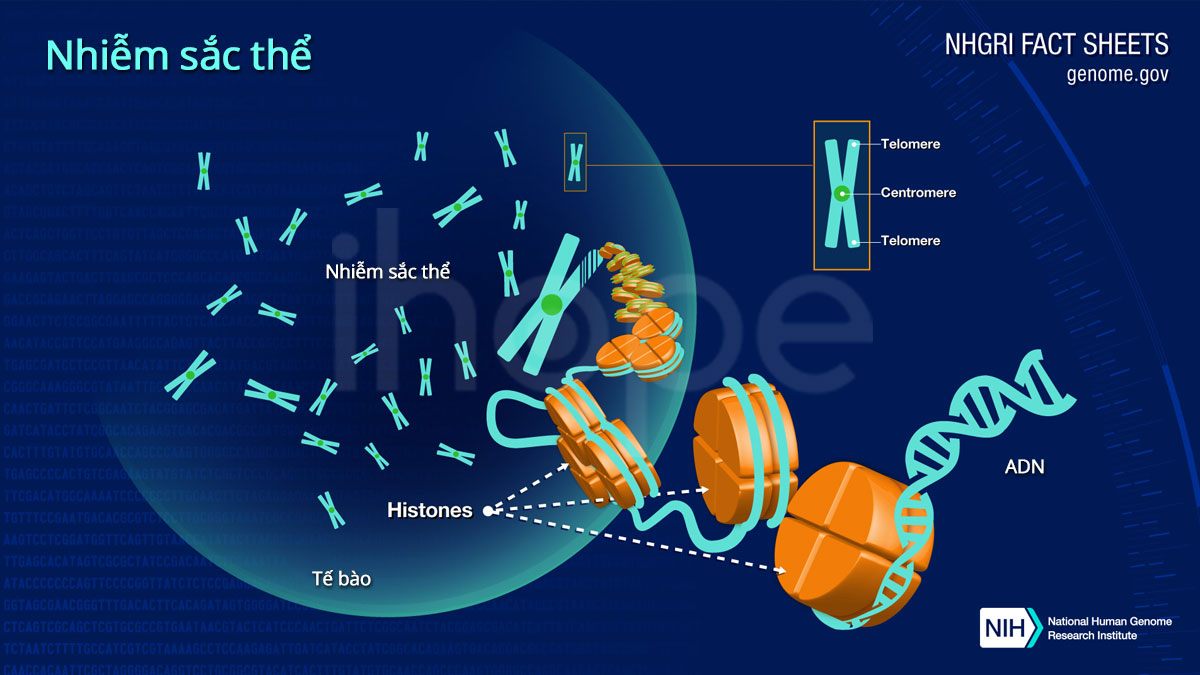
Nhiễm sắc thể 15 liên quan đến hội chứng Angelman, hội chứng Prader-Willi, bệnh bạch cầu nguyên bào cấp tính,… Người bình thường có 46 nhiễm sắc thể trong mỗi tế bào, được chia thành 23 cặp. Trong hai bản sao của nhiễm sắc thể số 15 thì mỗi bản sao được thừa hưởng từ mỗi bên cha và mẹ, tạo thành một trong các cặp. Nhiễm sắc thể 15 trải dài hơn 102 triệu cặp base cấu tạo nên ADN và chiếm hơn 3% tổng số ADN trong tế bào.
Tổng quan
Xác định gen trên mỗi nhiễm sắc thể là lĩnh vực nghiên cứu di truyền đầy khả thực. Bởi vì các nhà nghiên cứu sử dụng các cách tiếp cận khác nhau để dự đoán số lượng gen trên mỗi nhiễm sắc thể dẫn đến số lượng gen ước tính sẽ khác nhau. Nhiễm sắc thể 15 có thể chứa 600 đến 700 gen mang thông tin tổng hợp nên protein. Các protein này thực hiện nhiều vai trò khác nhau trong cơ thể.
Đột biến gây bệnh
Các tình trạng bệnh sau đây có nguyên nhân do đột biến liên quan đến thay đổi cấu trúc nhiễm sắc thể hoặc thay đổi số lượng bản sao của nhiễm sắc thể số 15.
Hội chứng lặp đoạn 15q11-q13
Lặp đoạn trên nhánh dài (q) của nhiễm sắc thể 15 có thể gây ra hội chứng lặp đoạn 15q11-q13 (hội chứng dup15q). Biểu hiện có thể bao gồm yếu cơ, thiểu năng trí tuệ, động kinh và rối loạn phổ tự kỷ.
Hội chứng lặp đoạn 15q xảy ra khi có thêm ít nhất một bản sao của vùng 15q11.2-q13.1 trên nhiễm sắc thể 15. Vùng này còn được gọi là vùng quan trọng Prader-Willi/Angelman (Prader-Willi/Angelman Critical Region - PWACR) vì nó cũng liên quan đến hội chứng Prader-Willi và hội chứng Angelman. Hội chứng dup15q chỉ phát sinh nếu bất thường xảy ra trên bản sao của nhiễm sắc thể được thừa hưởng từ mẹ. Mỗi người thừa hưởng một bản sao nhiễm sắc thể số 15 từ cha và mẹ. Tuy nhiên, một số gen trên nhiễm sắc thể này bao gồm cả gen trong vùng 15q11.2-13.1 chỉ hoạt động trên bản sao từ mẹ. Cơ chế kích hoạt gen có nguồn gốc từ cha mẹ là kết quả của một hiện tượng gọi là in dấu bộ gen (genomic imprinting).
Nhiễm sắc thể đẳng tâm 15 là dạng bất thường nhiễm sắc thể hay gặp nhất dẫn đến lặp đoạn 15q11.2-q13.1, nó xảy ra ở khoảng 80% trường hợp. Một nhiễm sắc thể đẳng tâm chứa các đoạn-phản-chiếu và có hai điểm thắt (tâm động) thay vì một tâm động như ở nhiễm sắc thể thường. Ở người có bộ nhiễm sắc thể đẳng tâm 15, tế bào có hai bản sao bình thường của nhiễm sắc thể 15 cộng với hai bản sao nhân đôi của đoạn ADN trên nhiễm sắc thể đẳng tâm, tổng cộng có bốn bản sao của đoạn đã nhân đôi.
Trong khoảng 20% các trường hợp mắc hội chứng dup15q, lặp đoạn xảy ra trên nhánh dài (q) của một trong hai bản sao nhiễm sắc thể số 15 trong mỗi tế bào. Hiện tượng này gọi là trùng lặp kẽ hở (interstitial duplication). Khi đó, các tế bào có hai bản sao của nhiễm sắc thể số 15, một trong số đó có thêm một bản sao của đoạn ADN, tạo thành tổng số ba bản sao của đoạn được lặpi.
Trong tất cả các trường hợp của hội chứng dup15q, vật liệu di truyền được sao chép dẫn đến có thêm bản sao của một số gen liên quan đến quá trình phát triển. Vật liệu di truyền bổ sung này phá vỡ sự phát triển bình thường, gây ra các biểu hiện đặc trưng của bệnh. Người bệnh thường có các dấu hiệu và triệu chứng nhẹ hơn so với những người mang nhiễm sắc thể đẳng tâm 15.
Mất đoạn 15q13.3
Mất đoạn 15q13.3 xảy ra khi một đoạn nhỏ của nhiễm sắc thể số 15 bị xóa trong mỗi tế bào. Mất đoạn xảy ra tại vị trí q13.3 trên nhánh q của nhiễm sắc thể 15. Hầu hết người bị mất đoạn 15q13.3 đều thiếu một trình tự khoảng 2 triệu cặp base ADN (2Mb). Kích thước đoạn bị xóa có thể xê dịch, nhưng nó thường chứa ít nhất sáu gen. Người ta chưa rõ bằng cách nào mà mất các gen này lại tăng nguy cơ thiểu năng trí tuệ, động kinh, các vấn đề hành vi và rối loạn tâm thần ở một số người bệnh.
Những người khác không có dấu hiệu hoặc triệu chứng rõ ràng do bị mất đoạn. Ở những cá nhân này, đột biến mất đoạn thường được phát hiện khi họ làm xét nghiệm di truyền vì có người thân mang bệnh. Người ta chưa rõ vì sao mất đoạn 15q13.3 lại gây ra rối loạn nhận thức và hành vi ở một số trường hợp, trong khi những người khác lại ít hoặc không có vấn đề về sức khỏe. Người ta cho rằng yếu tố di truyền hoặc môi trường có thể liên quan đến mức độ biểu hiện bệnh.
Mất đoạn 15q24
Đột biến mất đoạn 15q24 xảy ra khi một đoạn nhỏ của nhiễm sắc thể số 15 bị xóa trong mỗi tế bào. Cụ thể hơn, người bệnh bị mất đoạn chứa từ 1,7Mb đến 6,1Mb ADN tại vị trí q24 trên nhiễm sắc thể 15. Kích thước đoạn bị mất có thể xê dịch, nhưng tất cả đều thiếu vùng 1,2Mb như nhau, bao gồm một số gen quan trọng đối với quá trình phát triển bình thường. Người ta vẫn chưa rõ làm thế nào mất các gen này lại dẫn đến thiểu năng trí tuệ, khuôn mặt dị biệt và các bất thường khác.
Bệnh bạch cầu nguyên bào cấp tính
Một loại ung thư máu gọi là bệnh bạch cầu nguyên bào cấp tính (acute promyelocytic leukemia) do chuyển vị giữa nhiễm sắc thể 15 và nhiễm sắc thể 17. Chuyển vị t(15;17) hợp nhất một phần gen PML từ nhiễm sắc thể 15 với một phần gen RARA từ nhiễm sắc thể 17. Dạng đột biến này gọi là đột biến soma, nó không di truyền mà phát sinh trong quá trình sống của một người và chỉ hiện diện trong tế bào ung thư. Chuyển vị t(15;17) là chuyển đoạn cân bằng vì các đoạn nhiễm sắc thể trao đổi tương đương nhau, không thêm hay bớt vật chất di truyền. Protein PML-RARα được tạo ra từ gen hợp nhất.
Protein PML-RARα hoạt động khác với các protein bình thường tạo ra từ gen PML và RARA. Gen PML trên nhiễm sắc thể 15 cung cấp hướng dẫn tạo ra một loại protein có chức năng như một chất ức chế khối u, nó ngăn chặn tế bào phát triển và phân chia quá nhanh hoặc mất kiểm soát. Protein PML ngăn chặn tế bào phát triển và phân chia cũng như kết hợp với các protein khác để kích hoạt quá trình tự hủy (apoptosis).
Gen RARA trên nhiễm sắc thể 17 cung cấp hướng dẫn tạo ra một yếu tố phiên mã gọi là thụ thể axit retinoic alpha (Retinoic Acid Receptor alpha - RARα). Yếu tố phiên mã là protein gắn vào một số vùng ADN để kiểm soát hoạt động của các gen tương ứng. Thông thường, protein RARα kiểm soát hoạt động của các gen quan trọng với quá trình biệt hóa của tế bào bạch cầu chưa trưởng thành vượt qua giai đoạn promyelocyte. Protein PML-RARα can thiệp vào chức năng bình thường của cả PML và protein RARα, từ đó khiến các tế bào máu bị mắc kẹt tại giai đoạn tiền tế bào rồi tăng sinh bất thường. Các tế bào sinh tủy dư thừa tích tụ trong tủy xương và các tế bào bạch cầu bình thường không thể hình thành, dẫn đến bệnh bạch cầu cấp tính nguyên bào tủy.
Hội chứng Angleman
Hội chứng Angelman do mất các gen hoạt động trong vùng (15q11-q13) của nhiễm sắc thể số 15 trong mỗi tế bào. Vùng này nằm trên nhánh q của nhiễm sắc thể 15 có chứa gen UBE3A. Khi gen UBE3A bị đột biến hoặc bị xóa, có thể dẫn đến đặc điểm thần kinh đặc trưng của hội chứng Angelman.
Mỗi người thừa hưởng một bản sao gen UBE3A từ cha và mẹ, cả hai bản sao này đều hoạt động trong nhiều mô của cơ thể. Tuy nhiên, trong một số vùng nhất định của não, chỉ bản sao từ mẹ mới hoạt động. Cơ chế kích hoạt gen có nguồn gốc từ cha mẹ là kết quả của một hiện tượng gọi là in dấu bộ gen (genomic imprinting). Nếu bản sao từ mẹ bị mất do thay đổi cấu trúc nhiễm sắc thể hoặc đột biến gen, sẽ không có bản sao hoạt động của gen UBE3A trong một số bộ phận của não.
Trong hầu hết trường hợp (khoảng 70%), hội chứng Angelman xảy ra do bản sao nhiễm sắc thể 15 từ mẹ bị mất đoạn chứa gen UBE3A. Bởi vì bản sao gen UBE3A thừa hưởng từ cha thường không hoạt động tại một số bộ phận của não, nhiễm sắc thể 15 từ mẹ bị mất đoạn nên không thể có bản sao gen UBE3A hoạt động tại những vùng não này.
Trong 3-7% các trường hợp mắc hội chứng Angelman, bệnh xảy ra khi một người thừa hưởng hai bản sao nhiễm sắc thể số 15 từ cha thay vì một bản sao từ cha hoặc mẹ. Hiện tượng này gọi là lưỡng bội từ cha (paternal uniparental disomy - UPD). Những người bị UPD cha mang nhiễm sắc thể 15 có hai bản sao của gen UBE3A, nhưng cả hai đều được di truyền từ cha, do đó chúng không hoạt động trong não.
Khoảng 10% trường hợp mắc hội chứng Angelman do đột biến gen UBE3A, 3% do khiếm khuyết trong vùng ADN kiểm soát hoạt hóa gen UBE3A và các gen khác trên bản sao nhiễm sắc thể 15 từ mẹ. Một số ít trường hợp hội chứng Angelman do chuyển vị nhiễm sắc thể hoặc do đột biến gen khác ngoài UBE3A. Những thay đổi di truyền này làm bất hoạt gen UBE3A một cách bất thường.
Xem thêm hội chứng AngelmanHội chứng Prader-Willi
Hội chứng Prader-Willi có nguyên nhân do mất các gen trong vùng 15q11-q13 trên nhánh q của nhiễm sắc thể số 15. Nó cũng là vùng bị ảnh hưởng ở những người mắc hội chứng Angelman mặc dù liên quan các gen khác nhau. Một người có thể mắc hội chứng Prader-Willi hoặc hội chứng Angelman, nhưng thường không thể mắc cả hai.
Mỗi người thừa hưởng một bản sao nhiễm sắc thể số 15 từ cha và mẹ. Một số gen trên nhiễm sắc thể này chỉ hoạt động trên bản sao được thừa hưởng từ cha. Cơ chế kích hoạt gen có nguồn gốc từ cha mẹ là kết quả của một hiện tượng gọi là in dấu bộ gen (genomic imprinting).
Trong khoảng 70% trường hợp, hội chứng Prader-Willi xảy ra khi vùng 15q11-q13 của nhiễm sắc thể 15 từ người cha bị xóa trong mỗi tế bào. Do đó, một số gen quan trọng trong vùng này bị thiếu vì bản sao từ cha đã bị xóa trong khi bản sao từ mẹ bị tắt (không hoạt động). Người ta đang xác định những gen bị thiếu có liên quan đến các biểu hiện đặc trưng của hội chứng Prader-Willi.
Trong khoảng 25% trường hợp, người mắc hội chứng Prader-Willi thừa hưởng hai bản sao nhiễm sắc thể số 15 từ mẹ thay vì một bản sao từ cha hoặc mẹ. Hiện tượng này được gọi là lưỡng bội từ mẹ (maternal UPD). Một người mang hai bản sao nhiễm sắc thể 15 từ mẹ sẽ không có bản sao hoạt động của một số gen trong vùng 15q11-13.
Một số ít trường hợp hội chứng Prader-Willi do chuyển vị nhiễm sắc thể. Hiếm khi bệnh xảy ra do đột biến hoặc vấn đề khác làm bất hoạt bất thường các gen trên bản sao nhiễm sắc thể số 15 từ cha.
Xem thêm hội chứng Prader-WilliĐiếc thần kinh giác quan và vô sinh nam
Điếc thần kinh giác quan và vô sinh nam (sensorineural deafness and male infertility) xảy ra do mất đoạn trên nhánh q của nhiễm sắc thể số 15. Các triệu chứng liên quan do mất nhiều gen trong vùng này. Kích thước đoạn bị xóa khác nhau giữa từng trường hợp. Mất gen STRC trên nhiễm sắc thể 15 chính là nguyên nhân gây ra mất thính giác, trong khi mất gen CATSPER2 làm bất thường tinh trùng, dẫn đến vô sinh nam. Người ta đang xác định xem liệu mất thêm các gen trong vùng bị xóa ảnh hưởng đến những người bị điếc thần kinh giác quan và vô sinh nam. như thế nào.
Các vấn đề khác
Những thay đổi khác về số lượng hoặc cấu trúc của nhiễm sắc thể 15 có thể gây ra thiểu năng trí tuệ, chậm tăng trưởng và phát triển, yếu cơ và khuôn mặt dị biệt. Những thay đổi này bao gồm thêm một bản sao của một phần nhiễm sắc thể 15 (tam bội một phần 15), một đoạn nhiễm sắc thể bị thiếu trong mỗi tế bào (đơn bội một phần 15) và cấu trúc hình tròn được gọi là nhiễm sắc thể vòng 15. Nhiễm sắc thể dạng vòng xảy ra khi nhiễm sắc thể bị đứt tại hai vị trí, sau đó phần đầu của các nhánh nhiễm sắc thể hợp nhất với nhau tạo thành cấu trúc vòng tròn.
References
- National Human Genome Research Institute. Chromosome Abnormalities. Retrieved October 14, 2020 from https://www.genome.gov/about-genomics/fact-sheets/Chromosome-Abnormalities-Fact-Sheet
- U.S. National Library of Medicine. Chromosome 15. Retrieved February 15, 2022 from https://medlineplus.gov/genetics/chromosome/15/