
Hạ betalipoprotein máu gia đình (familial hypobetalipoproteinemia) là bệnh di truyền khiến cơ thể giảm khả năng hấp thụ và vận chuyển chất béo. Người bệnh có biểu hiện chung là nồng độ cholesterol trong máu thấp.
Nguồn: Naeblys/Shutterstock.com
Biểu hiện lâm sàng
Triệu chứng của bệnh hạ betalipoprotein máu gia đình có nhiều mức độ khác nhau.
Mức độ nhẹ
Phần lớn người bệnh mức độ nhẹ biểu hiện liên quan đến quá trình hấp thụ chất béo từ chế độ ăn và không có biểu hiện khác. Một số bệnh nhân xuất hiện triệu chứng tích trữ chất béo trong gan dẫn đến bệnh gan nhiễm mỡ.
Nguồn: Medlineplus.gov
Mức độ nghiêm trọng
Đối với trường hợp nghiêm trọng, bệnh gan nhiễm mỡ tiến triển thành bệnh gan mãn tính hay bệnh xơ gan. Người bệnh hạ betalipoprotein máu gia đình nghiêm trọng thường khó hấp thụ chất béo cũng như các vitamin tan trong chất béo như vitamin E và vitamin A. Từ đó, bệnh nhân xuất hiện dấu hiệu thừa chất béo trong phân (phân mỡ). Đối với trẻ nhỏ, vấn đề tiêu hóa khiến trẻ khó phát triển và không tăng cân như mong đợi (suy dinh dưỡng).

Nguồn: Cleveland Clinic
Độ phổ biến
Hiện nay, tỉ lệ mắc bệnh hạ betalipoprotein máu gia đình vào khoảng 1/1.000–1/3.000 người.
Nguyên nhân
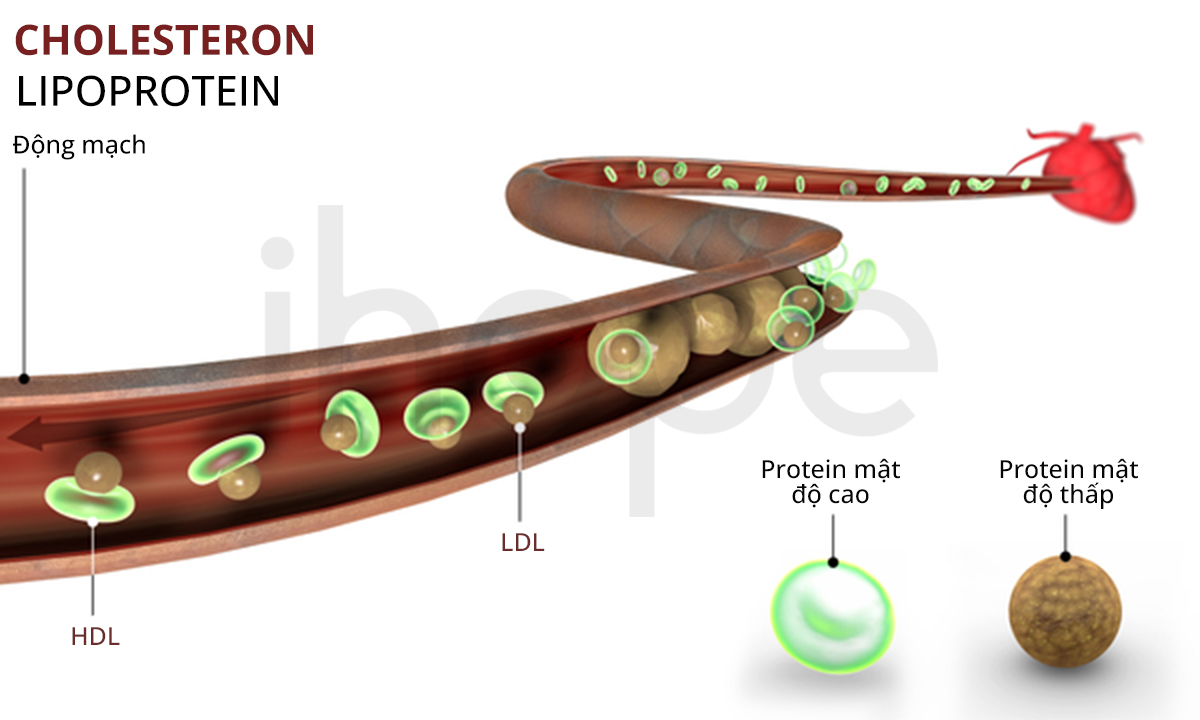
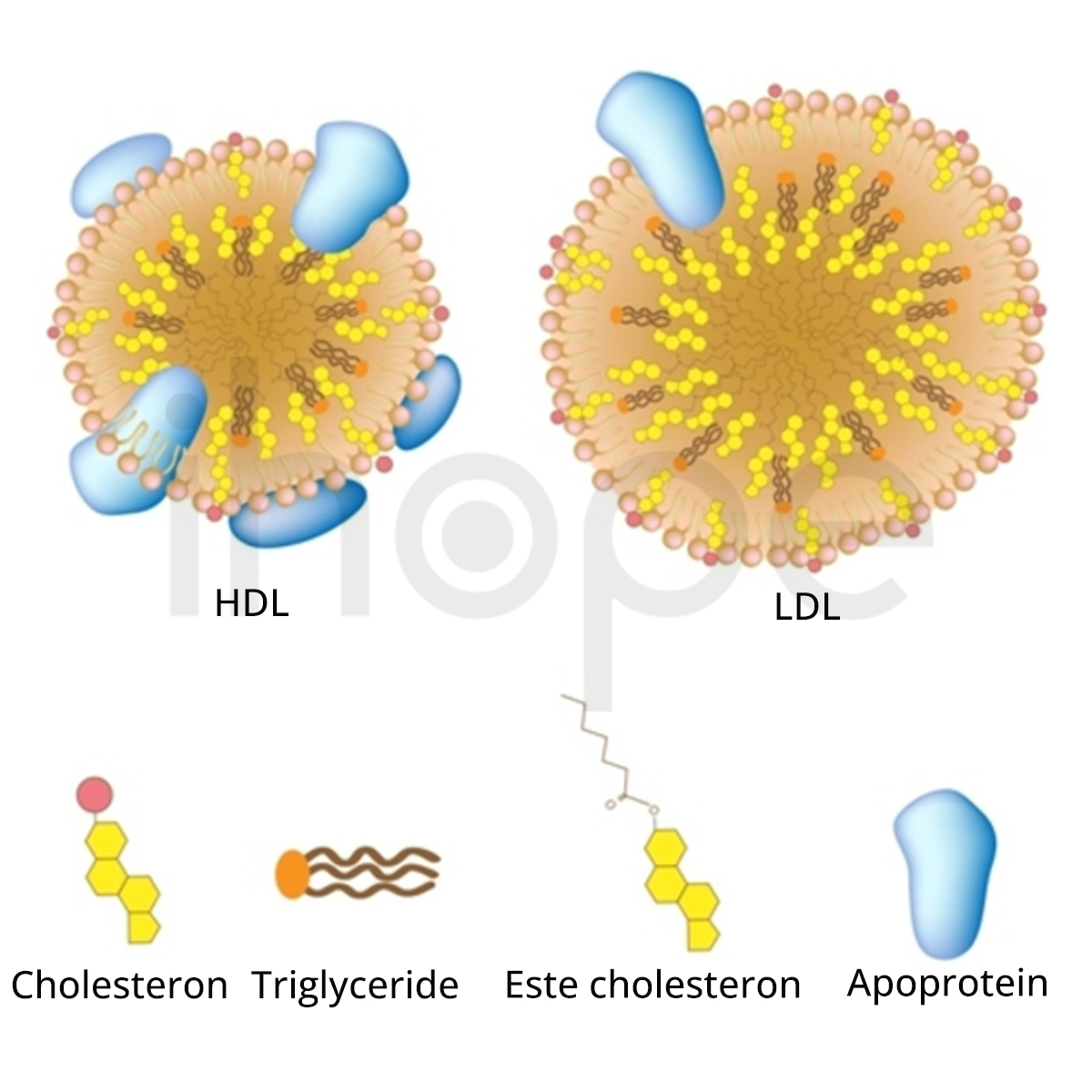
Đột biến gen APOB gây ra bệnh hạ betalipoprotein máu gia đình. Gen APOB cung cấp hướng dẫn tạo ra hai dạng của protein apolipoprotein B gồm dạng ngắn (apolipoprotein B-48) và dạng dài (apolipoprotein B-100). Cả hai dạng này đều là thành phần của lipoprotein. Lipoprotein giữ vai trò vận chuyển chất béo và cholesterol trong máu.
Nguồn: U.S. National Library of Medicine
Đa số đột biến gen APOB khiến hai dạng của apolipoprotein B trở nên ngắn hơn so với bình thường. Mức độ nghiêm trọng của bệnh chủ yếu phụ thuộc vào độ dài của hai dạng apolipoprotein B. Dạng siêu ngắn sẽ không thể kết hợp với lipoprotein để vận chuyển chất béo và cholesterol. Protein ngắn hơn bình thường một chút vẫn có khả năng hoạt động nhưng hiệu quả kết hợp với lipoprotein không cao. Nhìn chung, những dấu hiệu của bệnh tiến triển nghiêm trọng hơn khi cả hai dạng của apolipoprotein B trở nên cực ngắn. Trong trường hợp bệnh nhẹ hoặc không có triệu chứng, protein thường chỉ ngắn hơn bình thường rất ít. Những thay đổi này khiến apolipoprotein B giảm chức năng và giảm hoặc mất khả năng vận chuyển chất béo cũng như cholesterol. Do đó, người bệnh giảm khả năng hấp thụ chất béo và vitamin tan trong chất béo.
Đa số người bệnh mang đột biến gen APOB. Trong một số ít trường hợp, bệnh nhân có đột biến tại các gen khác. Ngoài ra, có những bệnh nhân không mang bất kì đột biến gen nào. Do đó, người ta cho rằng nguyên nhân còn có thể bắt nguồn từ những gen chưa được xác định.
Chẩn đoán
Bác sĩ chẩn đoán hạ betalipoprotein máu gia đình dựa trên những đánh giá lâm sàng, xét nghiệm sinh hóa. Các xét nghiệm sinh hóa gồm xét nghiệm nồng độ LDL-C và apolipoprotein B trong huyết tương. Ngoài ra, đánh giá bệnh sử gia đình cũng cần thiết cho quá trình chẩn đoán. Mặt khác, bệnh nhân có thể thực hiện xét nghiệm di truyền nhằm xác nhận chẩn đoán.
Điều trị
Hiện nay, chưa có liệu pháp điều trị hoàn toàn bệnh hạ betalipoprotein máu gia đình. Các liệu pháp tập trung làm giảm triệu chứng và cải thiện cuộc sống cho người bệnh.
Các phương pháp điều trị phổ biến gồm có:
- Chế độ ăn cung cấp lượng calo hấp thụ cần thiết và lượng triglyceride hợp lí
- Hạn chế hấp thụ chất béo không cần thiết
- Bổ sung axit béo thiết yếu, sắt, các loại vitamin A, D, E, K, B9 và B12 qua đường uống
- Phẫu thuật ghép gan
- Hỗ trợ đa chuyên khoa cho từng biến chứng
Dạng di truyền
Hạ betalipoprotein máu gia đình di truyền theo kiểu trội trên nhiễm sắc thể thường. Do đó, một bản sao của gen trong mỗi tế bào đủ để gây ra bệnh. Người bệnh thường được di truyền từ cha hoặc mẹ mắc bệnh. Một số ít trường hợp do đột biến mới (denovo) xuất hiện trong quá trình hình thành tế bào sinh sản (trứng, tinh trùng) hoặc quá trình phát triển phôi sớm.
Đối với bệnh này, một thay đổi tại một bản sao của gen APOB trong mỗi tế bào sẽ gây ra bệnh. Khi đột biến xảy ra trên cả hai bản sao của gen sẽ gây nên nhiều vấn đề sức khỏe với mức độ nghiêm trọng hơn.

Nguồn: U.S. National Library of Medicine
Phòng ngừa
Hạ betalipoprotein máu gia đình di truyền trội trên nhiễm sắc thể thường, nếu cha hoặc mẹ mắc bệnh, con sinh ra sẽ có 50% khả năng di truyền bệnh. Do đó, để bảo đảm 100% khả năng con không bị bệnh, cha mẹ có thể chọn phương pháp thụ tinh nhân tạo IVF và sàng lọc phôi PGS/PGD. Các thành viên trong gia đình nên khám sức khỏe và tầm soát bệnh định kỳ nếu có thành viên mắc bệnh. Các cặp vợ chồng trước khi mang thai cần tư vấn và xét nghiệm di truyền> nhằm đảm bảo sinh con khỏe mạnh.
Các tên gọi khác
- FHBL
- Hypobetalipoproteinemia
References
- Genetic Testing Information. Familial hypobetalipoproteinemia. Retrieved August 13, 2024 from https://www.ncbi.nlm.nih.gov/gtr/conditions/C1862596/
- Genetic and Rare Diseases Information Center. Hypobetalipoproteinemia, familial, 1. Retrieved August 13, 2024 from https://rarediseases.info.nih.gov/diseases/2876/index
- Catalog of Genes and Diseases from OMIM. HYPOBETALIPOPROTEINEMIA, FAMILIAL, 2; FHBL2. Retrieved August 13, 2024 from https://omim.org/entry/605019
- U.S. National Library of Medicine. Familial hypobetalipoproteinemia. Retrieved August 13, 2024 from https://medlineplus.gov/genetics/condition/familial-hypobetalipoproteinemia/
- Orphanet. Hypobetalipoproteinemia. Retrieved August 13, 2024 from https://www.orpha.net/en/disease/detail/31154
- National Institute of Health. APOB-Related Familial Hypobetalipoproteinemia. Retrieved August 13, 2024 from https://www.ncbi.nlm.nih.gov/books/NBK570370/
- National Organization for Rare Disorders. familial hypobetalipoproteinemia 1. Retrieved August 13, 2024 from https://rarediseases.org/mondo-disease/familial-hypobetalipoproteinemia-1/
- American Heart Association. Hepatic and Cardiovascular Consequences of Familial Hypobetalipoproteinemia. Retrieved August 13, 2024 from https://www.ahajournals.org/doi/full/10.1161/01.atv.0000176191.64314.07
- AACE Clinical Case Reports. Hypolipidemia due to Familial Hypobetalipoproteinemia in Adolescents. Retrieved August 13, 2024 from https://www.aaceclinicalcasereports.com/article/S2376-0605(24)00032-4/fulltext





