
Thiếu dihydrolipoamide dehydrogenase (dihydrolipoamide dehydrogenase deficiency) là bệnh di truyền tác động đến nhiều hệ thống trong cơ thể. Những dấu hiệu của bệnh thường xuất hiện ngay sau khi trẻ sinh ra với nhiều mức độ nghiêm trọng.
Biểu hiện lâm sàng
Biểu hiện phổ biến
Người bệnh thiếu dihydrolipoamide dehydrogenase có triệu chứng phổ biến sau:
- Buồn nôn, nôn
- Hô hấp khó khăn
- Nhịp tim không ổn định
Tình trạng axit lactic tích tụ là nguyên nhân chính dẫn đến các biểu hiện trên. Triệu chứng của bệnh xuất hiện theo từng đợt do ảnh hưởng của những yếu tố như sốt, chấn thương và căng thẳng. Giữa mỗi đợt tái phát, người bệnh không xuất hiện bất kì dấu hiệu nào. Trẻ sơ sinh mắc bệnh thường không sống sót qua vài năm đầu đời do mức độ nghiêm trọng của bệnh. Bệnh nhân sống sót qua giai đoạn trẻ nhỏ thường phát triển chậm và gặp nhiều vấn đề liên quan đến thần kinh như thiểu năng trí tuệ, cứng cơ, rối loạn vận động và co giật.
Thần kinh
Ngoài ra, trẻ sơ sinh thiếu dihydrolipoamide dehydrogenase còn xuất hiện các biểu hiện liên quan đến thần kinh bao gồm:
- Giảm trương lực cơ
- Hôn mê
Mức độ nghiêm trọng của những triệu chứng này tăng dần khiến trẻ gặp khó khăn khi bú, mất tỉnh táo và co giật.
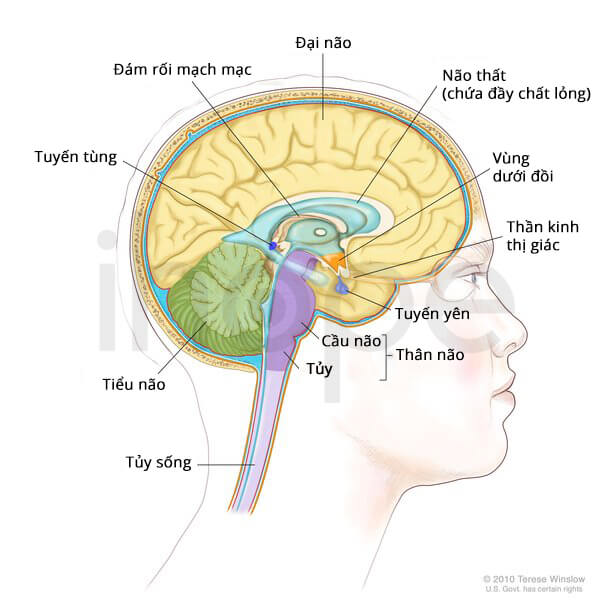
Nguồn: Terese Winslow
Gan
Thiếu dihydrolipoamide dehydrogenase còn gây ra triệu chứng liên quan đến gan với mức độ nhẹ như gan phình to. Nếu không điều trị kịp thời, bệnh sẽ phát triển thành suy gan, nghiêm trọng hơn, bệnh có thể đe dọa đến tính mạng của trẻ.

Nguồn: Mount Sinai
Trong một số trường hợp, bệnh nhân chỉ xuất hiện biểu hiện về gan. Bệnh gan có thể khởi phát bất kì lúc nào từ giai đoạn trẻ nhỏ đến khi trưởng thành. Nó khiến người bệnh nôn mửa và đau bụng tái phát.
Trong một số trường hợp, người bệnh xuất hiện các biểu hiện khác như:
- Yếu cơ xương khi vận động và tập thể dục
- Mí mắt sụp
- Suy cơ tim
- Hàm lượng amoniac trong máu tăng
- Tích lũy ketone trong cơ thể
- Hạ đường huyết
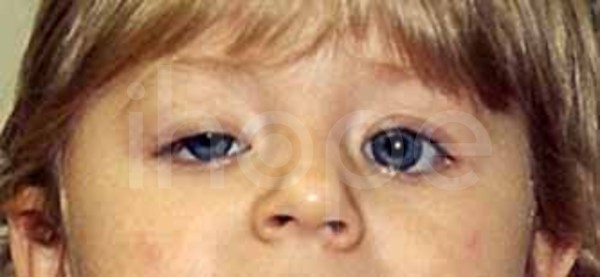
Ảnh: Mí mắt sụp xuống
Nguồn: Elements of Morphology, National Human Genome Research Institute
Độ phổ biến
Hiện nay, tỉ lệ mắc bệnh thiếu dihydrolipoamide dehydrogenase khoảng 1/48.000–1/35.000 người gốc Do Thái Ashkenazi. Nhóm người này chủ yếu xuất hiện triệu chứng liên quan đến gan. Đối với quần thể khác, tỉ lệ thiếu dihydrolipoamide dehydrogenase chưa được xác định. Tuy nhiên, người ta cho rằng bệnh này rất hiếm xảy ra.
Nguyên nhân
Đột biến gen DLD gây ra thiếu dihydrolipoamide dehydrogenase. Gen DLD cung cấp hướng dẫn tạo ra enzyme dihydrolipoamide dehydrogenase (DLD).
Enzyme dihydrolipoamide dehydrogenase là thành phần của ba nhóm enzyme sau hoạt động cùng nhau:
- Alpha-keto axit dehydrogenase phân nhánh (BCKD): phân giải ba loại axit amin phổ biến trong thực phẩm giàu protein bao gồm leucine, isoleucine và valine nhằm cung cấp phân tử năng lượng cho tế bào.
- Pyruvate dehydrogenase (PDH): tham gia vào quá trình chuyển hóa năng lượng từ thực phẩm thành năng lượng đơn giản để tế bào sử dụng.
- Alpha-ketoglutarate dehydrogenase (αKGDH): có chức năng tương tự enzyme pyruvate dehydrogenase—tham gia vào quá trình biến đổi năng lượng từ thức ăn để cung cấp cho cơ thể.
Đột biến gen DLD làm giảm chức năng của enzyme dihydrolipoamide dehydrogenase và ngăn cản phức hợp 3 enzyme thực hiện chức năng của chúng. Do đó, những phân tử và sản phẩm phụ của quá trình phân giải dần tích tụ trong cơ thể đến mức làm tổn thương mô, gây nhiễm axit lactic và mất cân bằng hóa học trong cơ thể. Đồng thời, năng lượng do tế bào tạo ra cũng giảm đáng kể.
Các quá trình bất thường này kết hợp với nhau tạo ra ảnh hưởng lớn đến não khiến người bệnh xuất hiện các dấu hiệu liên quan đến thần kinh. Mặt khác, tế bào giảm sản xuất năng lượng cũng tác động tiêu cực đến gan . Mức độ nghiêm trọng của bệnh phụ thuộc vào độ suy giảm chức năng của các phức hợp.
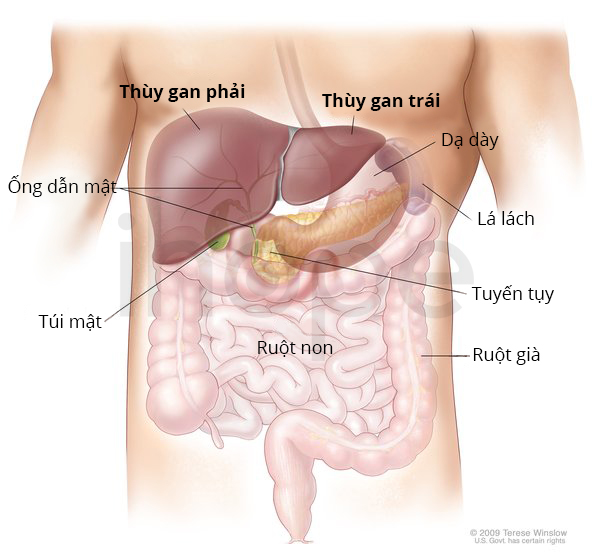
Ảnh: Giải phẫu gan
Nguồn: Medlineplus.gov
Chẩn đoán
Bác sĩ thường chẩn đoán thiếu dihydrolipoamide dehydrogenase dựa trên những dấu hiệu lâm sàng của bệnh. Ngoài ra, bác sĩ có thể chỉ định bệnh nhân xét nghiệm di truyền nhằm xác nhận kết quả chẩn đoán.
Xét nghiệm di truyền phổ biến gồm có:
- Xét nghiệm đơn gen nhằm xác định các đột biến trên những đoạn gen nhỏ như thêm, mất, lặp đoạn, đột biến vô nghĩa hoặc đột biến lệch khung
- Xét nghiệm đa gen nhằm xác định đột biến trên gen DLC và những gen khác liên quan đến bệnh
Điều trị
Hiện nay, chưa có liệu pháp điều trị hoàn toàn thiếu dihydrolipoamide dehydrogenase. Các liệu pháp tập trung làm giảm triệu chứng bệnh và cải thiện cuộc sống cho người bệnh.
Liệu pháp điều trị hằng ngày
Liệu pháp điều trị hằng ngày dành cho những người bệnh khởi phát sớm thường tập trung vào điều chỉnh lượng protein hấp thụ. Người bệnh nên có chế độ ăn giàu chất béo/ketogenic, bổ sung thêm dichloroacetate. Đối với bệnh nhân có lượng bạch cầu tăng đáng kể nên hấp thụ những protein không phân nhánh. Ngoài ra, người bệnh nên thực hiện thêm các liệu pháp điều trị khác nếu có dấu hiệu thiểu năng trí tuệ, rối loạn chức năng tim và giảm thị giác.
Điều trị nội trú cấp tính
Các phương pháp điều trị nội trú cấp tính được áp dụng cho bệnh nhân có triệu chứng thần kinh bao gồm:
- Sử dụng dung dịch muối và chất điện giải
- Điều trị bằng bicarbonate khi bệnh nhân nhiễm toan nghiêm trọng
- Kiểm tra lượng protein hấp thụ
Đối với những bệnh nhân có dấu hiệu liên quan đến gan, bác sĩ sẽ áp dụng các phương pháp sau:
- Truyền dịch tĩnh mạch chứa dextrose và chất điện giải
- Điều trị nhiễm toan bằng natri bicarbonate
- DCA hoặc thẩm phân máu
- Sử dụng huyết tương đông lạnh để điều trị triệu chứng máu khó đông
Người xuất hiện dấu hiệu liên quan đến cơ nên cân nhắc điều trị bằng riboflavin (220 mg/ngày).
Dạng di truyền
Thiếu dihydrolipoamide dehydrogenase truyền theo kiểu lặn trên nhiễm sắc thể thường, nghĩa là cả hai bản sao của gen trong mỗi tế bào đều có đột biến. Cha mẹ của một cá nhân mắc bệnh lặn trên nhiễm sắc thể thường đều mang một bản sao của gen đột biến, nhưng họ thường không biểu hiện dấu hiệu và triệu chứng của bệnh.

Nguồn: U.S. National Library of Medicine
Phòng ngừa
Thiếu dihydrolipoamide dehydrogenase di truyền lặn do đột biến gen DLD, cha mẹ mang đột biến dị hợp nên gần như không có biểu hiện bệnh, do đó rất khó phát hiện cho đến khi sinh con. Cha mẹ nên làm xét nghiệm sàng lọc gen lặn để chủ động cho tương lai của con. Các cặp vợ chồng trước khi mang thai cần tư vấn và xét nghiệm di truyền nhằm đảm bảo sinh con khỏe mạnh. Người thuộc nhóm nguy cơ mắc bệnh cần khám sức khỏe và tầm soát bệnh định kì.
Các tên gọi khác
- GSD type VI
- GSD VI
- GSD6
- Hepatic glycogen phosphorylase deficiency
- Hers disease
- Liver phosphorylase deficiency syndrome
References
- Genetic and Rare Diseases Information Center. Pyruvate dehydrogenase E3 deficiency. Retrieved July 29, 2024 from https://rarediseases.info.nih.gov/diseases/3263/index
- Catalog of Genes and Diseases from OMIM. DIHYDROLIPOAMIDE DEHYDROGENASE DEFICIENCY; DLDD. Retrieved July 29, 2024 from https://omim.org/entry/246900
- U.S. National Library of Medicine. Dihydrolipoamide dehydrogenase deficiency. Retrieved July 29, 2024 from https://medlineplus.gov/genetics/condition/dihydrolipoamide-dehydrogenase-deficiency/
- Metabolic Support UK. Dihydropyrimidine Dehydrogenase Deficiency. Retrieved July 29, 2024 from https://metabolicsupportuk.org/condition/dihydropyrimidine-dehydrogenase-deficiency-2/
- National Organization for Rare Disorders. Dihydrolipoamide dehydrogenase deficiency. Retrieved July 29, 2024 from https://rarediseases.org/gard-rare-disease/dihydrolipoamide-dehydrogenase-deficiency/
- Jscreen. Lipoamide Dehydrogenase Deficiency. Retrieved July 29, 2024 from https://www.jscreen.org/learn-more/diseases/dihydrolipoamide-dehydrogenase-deficiency/
- MalaCards. Dihydrolipoamide Dehydrogenase Deficiency (DLDD). Retrieved July 29, 2024 from https://www.malacards.org/card/dihydrolipoamide_dehydrogenase_e3_deficiency





