Thiếu lipoprotein lipase gia đình (familial lipoprotein lipase deficiency) là bệnh di truyền làm gián đoạn quá trình phân giải chất béo trong cơ thể dẫn đến một số loại chất béo tích tụ với hàm lượng lớn trong cơ thể.
Biểu hiện lâm sàng
Người bệnh thiếu lipoprotein lipase gia đình thường xuất hiện dấu hiệu trước 10 tuổi, trong số đó, tỉ lệ xuất hiện dấu hiệu trước 1 tuổi vào khoảng 25%.
Tụy
Triệu chứng khởi phát đầu tiên là đau bụng với nhiều mức độ từ nhẹ đến nghiêm trọng. Thông thường, nguyên nhân đau bụng đến từ viêm tụy cấp—cơn đau cấp tính đột ngột. Nếu không điều trị kịp thời, viêm tụy cấp sẽ tiến triển thành dạng mãn tính, gây tổn thương tụy và đe dọa đến tính mạng.
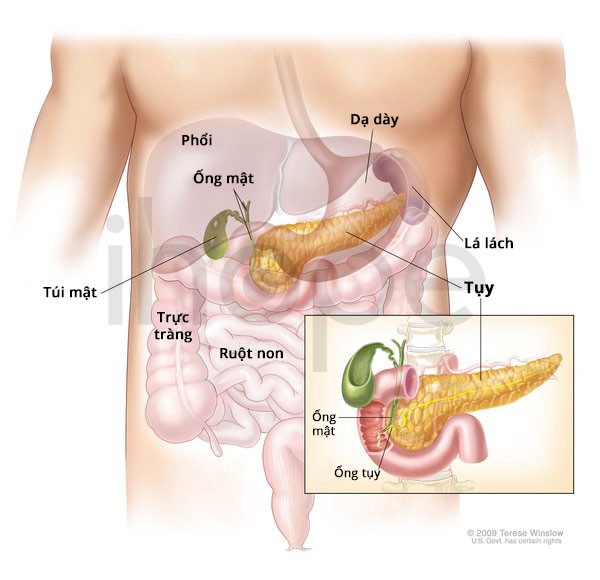
Nguồn: Terese Winslow LLC
Gan và lá lách
Người bệnh có kích thước gan và lá lách lớn. Mức độ phình to của gan và lá tỉ lệ thuận với nồng độ chất béo trong cơ thể. Khi lượng chất béo tăng, nhiều bạch cầu như đại thực bào sẽ tăng cường hấp thụ chất béo dư thừa nhằm giảm lượng chất béo trong máu. Sau khi hấp thụ chất béo, đại thực bào di chuyển về gan và lá lách—nơi tế bào mỡ tích tụ.
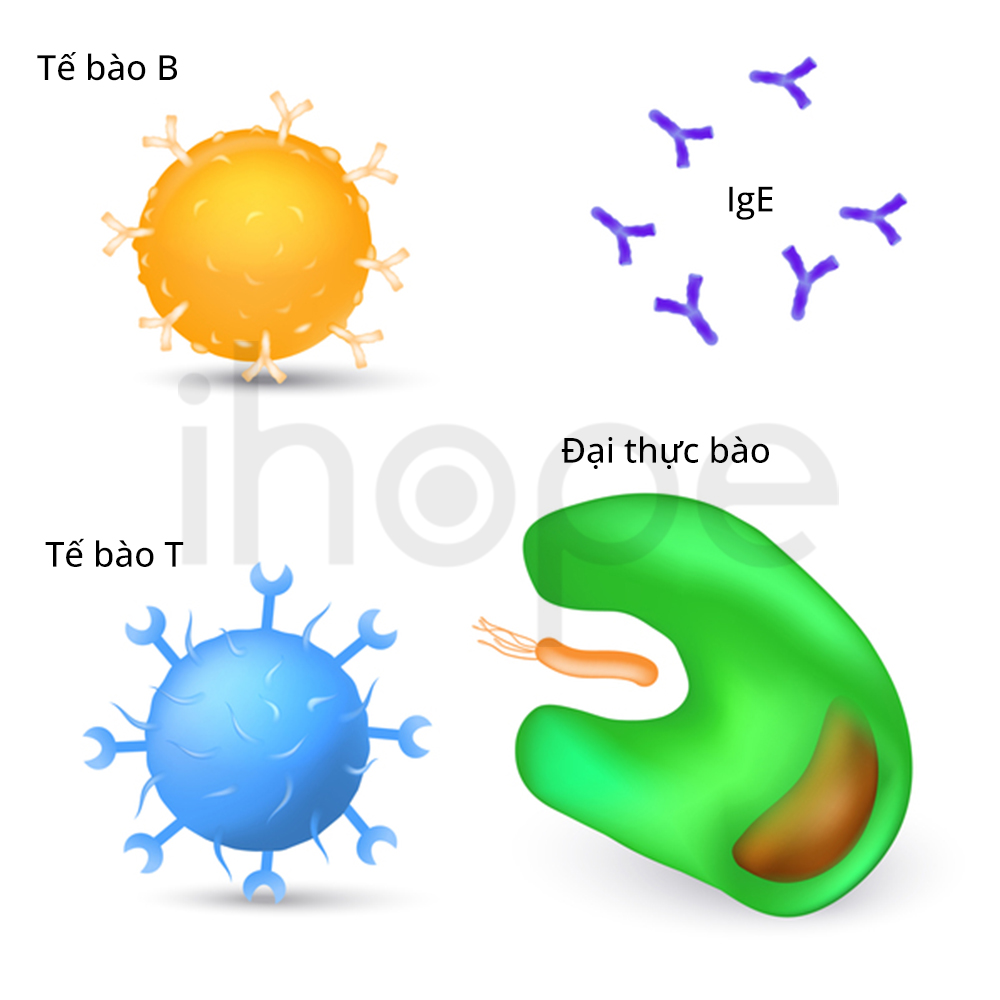
Ảnh: Tế bào B, tế bào T, đại thực bào và kháng thể
Nguồn: Designua/Shutterstock.com
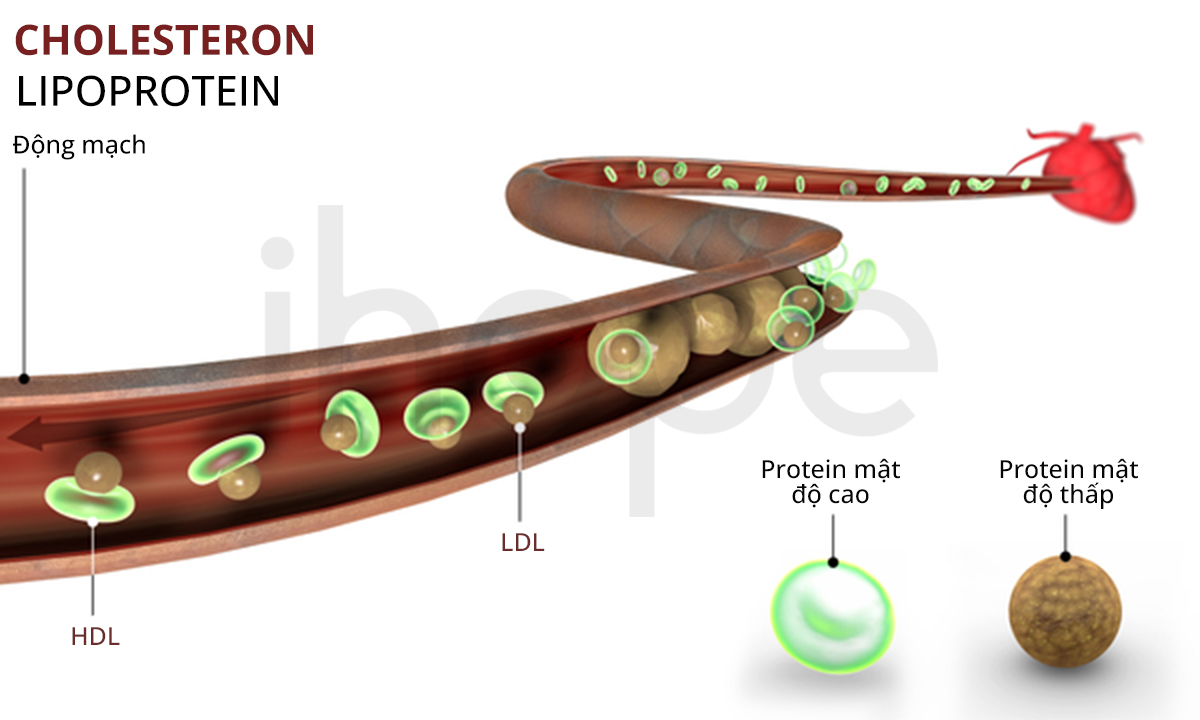
Nguồn: Naeblys/Shutterstock.com
U vàng phát ban
Khoảng một nửa bệnh nhân có biểu hiện u vàng phát ban—tổn thương nhỏ xuất hiện trên da do axit béo lắng động. Những mảng mỡ này thường xuất hiện trên thân, mông, đầu gối và cánh tay. U vàng phát ban có kích thước nhỏ (đường kính khoảng 1 mm) nhưng chúng có thể tập trung lại với nhau tạo thành các mảng lớn. Bình thường, những mảng này không gây đau đớn trừ khi có ma sát hoặc tác động lặp lại nhiều lần. U vàng phát ban thường bắt đầu xuất hiện khi lượng chất béo nạp vào tăng lên và nồng độ tích tụ cao; ngược lại, những mảng này biến mất khi lượng chất béo hấp thụ giảm và mức độ tích tụ thấp.
Mắt
Máu của người bệnh có màu trắng đục do hàm lượng chất béo cao. Chất béo thường tích tụ trong các mạch máu tại các mô lót phía sau võng mạc khiến mô này có màu hồng nhạt—bệnh nhiễm lipid võng mạc. Quá trình tích tụ không ảnh hưởng đến thị lực và thường biến mất khi người bệnh điều chỉnh chế độ ăn và lượng chất béo trong cơ thể giảm.
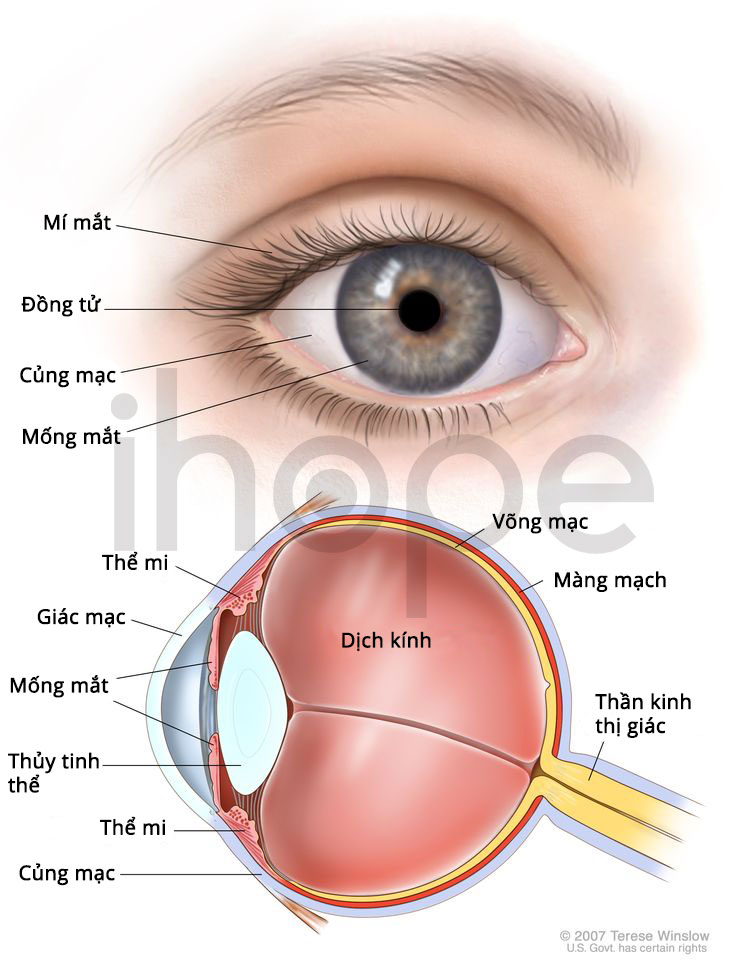
Nguồn: Medlineplus.gov
Thần kinh
Bệnh nhân còn xuất hiện dấu hiệu liên quan đến thần kinh như trầm cảm, mất trí nhớ và suy giảm trí tuệ nhẹ. Các triệu chứng này có thể được khắc phục khi tỉ lệ chất béo trong thức ăn giảm.
Độ phổ biến
Hiện nay, tỉ lệ mắc bệnh thiếu lipoprotein lipase gia đình vào khoảng 1/1.000.000 người trên toàn thế giới. Bệnh phổ biến tại Quebec, Canada.
Nguyên nhân
Đột biến gen LPL gây ra thiếu lipoprotein lipase gia đình. Gen LPL cung cấp hướng dẫn tạo ra enzyme lipoprotein lipase. Enzyme này chủ yếu tập trung trên bề mặt tế bào lót mao mạch trong mô cơ và mô mỡ. Enzyme lipoprotein lipase có chức năng phân giải chất béo triglyceride do các phân tử lipoprotein vận chuyển.
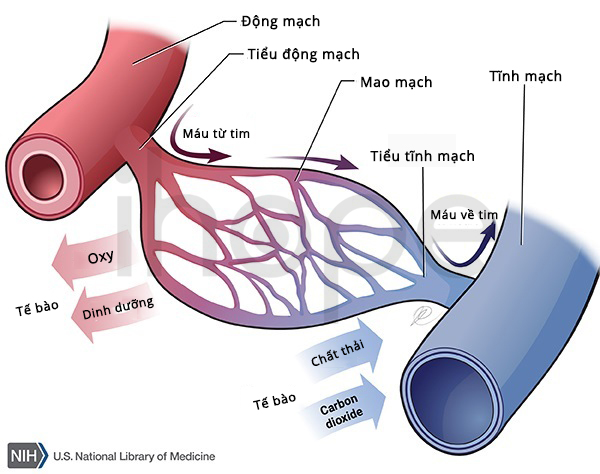
Nguồn: U.S. National Library of Medicine
Đột biến gen LPL khiến lipoprotein lipase giảm hoạt động hoặc mất chức năng nên quá trình phân giải triglyceride suy giảm. Cuối cùng, lượng triglyceride gắn với lipoprotein tích tụ trong máu và mô dẫn đến các dấu hiệu lâm sàng của bệnh thiếu lipoprotein lipase gia đình.
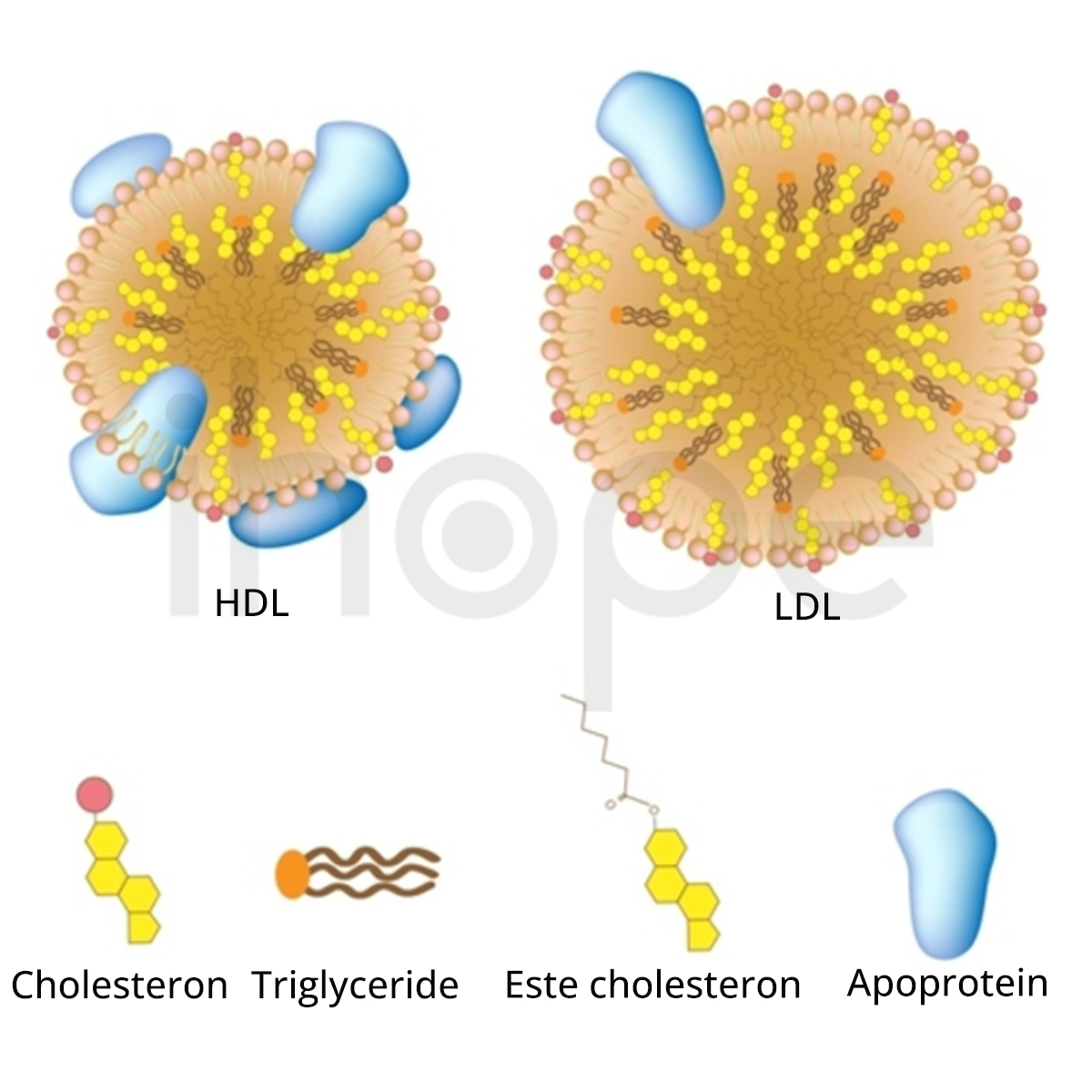
Nguồn: U.S. National Library of Medicine
Chẩn đoán
Bác sĩ chẩn đoán thiếu lipoprotein lipase gia đình dựa trên những triệu chứng đặc trưng và bệnh sử gia đình. Ngoài ra, bác sĩ còn chỉ định thực hiện xét nghiệm máu nhằm kiểm tra chức năng của enzyme lipoprotein lipase khi tiêm heparin vào tĩnh mạch. Heparin có khả năng kích thích gan tiết ra enzyme lipoprotein lipase. Ngoài ra, bệnh nhân có thể yêu cầu thực hiện xét nghiệm di truyền để xác nhận kết quả chẩn đoán.
Điều trị
Đa số bệnh nhân thiếu lipoprotein lipase gia đình điều trị thành công bằng cách hạn chế chất béo trong chế độ ăn. Tuy nhiên, một số người bệnh vẫn tái phát bệnh viêm tụy cấp và đau bụng. Mục đích của liệu pháp điều trị là hạn chế lượng chất béo hấp thụ nhưng vẫn duy trì lượng triglyceride cần thiết.
Thuốc làm giảm lượng chất béo trong cơ thể không hiệu quả với người bệnh. Người bệnh cần tránh hấp thụ cồn và thuốc làm tăng lượng triglyceride như thuốc uống tránh thai, thuốc lợi tiểu, thuốc ức chế beta–adrenergic, isotrtinoin và Zoloft. Bên cạnh đó, người bệnh nên có chế độ ăn giàu chất béo trung bình. Các thực phẩm chức năng từ dầu cá thường không mang lại hiệu quả cao và chống chỉ định sử dụng.
Khi mức triglyceride trong cơ thể giảm, kích thước của gan và lá lách sẽ trở lại bình thường trong vòng một tuần. U vàng phát ban cũng biến mất sau vài tuần hoặc vài tháng. Nếu u vàng vẫn tồn tại hoặc tái phát dù đã điều trị, người bệnh có thể đã không tuân thủ đầy đủ liệu pháp.
Dạng di truyền
Thiếu lipoprotein lipase gia đình di truyền theo kiểu lặn trên nhiễm sắc thể thường, nghĩa là cả hai bản sao của gen trong mỗi tế bào đều có đột biến. Cha mẹ của một cá nhân mắc bệnh lặn trên nhiễm sắc thể thường đều mang một bản sao của gen đột biến, nhưng họ thường không biểu hiện dấu hiệu và triệu chứng của bệnh.
Thành viên trong gia đình của người bệnh mang một đột biến trên bản sao của gen LPL trong mỗi tế bào, lượng chất béo trong máu của họ có thể tăng nhẹ. Do đó, họ tăng nguy cơ mắc các vấn đề sức khỏe như bệnh tim hoặc bệnh tiểu đường.

Nguồn: U.S. National Library of Medicine
Phòng ngừa
Thiếu lipoprotein lipase gia đình di truyền lặn do đột biến gen LPL, cha mẹ mang đột biến dị hợp nên gần như không có biểu hiện bệnh, do đó rất khó phát hiện cho đến khi sinh con. Cha mẹ nên làm xét nghiệm sàng lọc gen lặn để chủ động cho tương lai của con. Các cặp vợ chồng trước khi mang thai cần tư vấn và xét nghiệm di truyền nhằm đảm bảo sinh con khỏe mạnh. Người thuộc nhóm nguy cơ mắc bệnh cần khám sức khỏe và tầm soát bệnh định kỳ.
Các tên gọi khác
- Burger-Grutz syndrome
- Endogenous hypertriglyceridaemia
- Familial fat-induced hypertriglyceridemia
- Familial hyperchylomicronemia
- Familial LPL deficiency
- Hyperlipoproteinemia type I
- Hyperlipoproteinemia type Ia
- Lipase D deficiency
- LIPD deficiency
- Lipoprotein lipase deficiency, familial
References
- Genetic Testing Information. Hyperlipoproteinemia, type I. Retrieved August 06, 2024 from https://www.ncbi.nlm.nih.gov/gtr/conditions/C0023817/
- Genetic and Rare Diseases Information Center. Familial lipoprotein lipase deficiency. Retrieved August 06, 2024 from https://rarediseases.info.nih.gov/diseases/12241/index
- Catalog of Genes and Diseases from OMIM. HYPERLIPOPROTEINEMIA, TYPE I. Retrieved August 06, 2024 from https://omim.org/entry/238600
- U.S. National Library of Medicine. Familial lipoprotein lipase deficiency. Retrieved August 06, 2024 from https://medlineplus.gov/genetics/condition/familial-lipoprotein-lipase-deficiency/
- National Institute of Health. Lipoprotein Lipase Deficiency. Retrieved August 06, 2024 from https://www.ncbi.nlm.nih.gov/books/NBK560795/
- National Organization for Rare Disorders. Familial Lipoprotein Lipase Deficiency. Retrieved August 06, 2024 from https://rarediseases.org/rare-diseases/familial-lipoprotein-lipase-deficiency/
- Orphanet. Familial lipoprotein lipase deficiency. Retrieved August 06, 2024 from https://www.orpha.net/en/disease/detail/309015
- MalaCards. Familial Lipoprotein Lipase Deficiency. Retrieved August 06, 2024 from https://www.malacards.org/card/familial_lipoprotein_lipase_deficiency
- Nature. 805 Familial Lipoprotein Lipase Deficiency. Retrieved August 06, 2024 from https://www.nature.com/articles/pr20101014