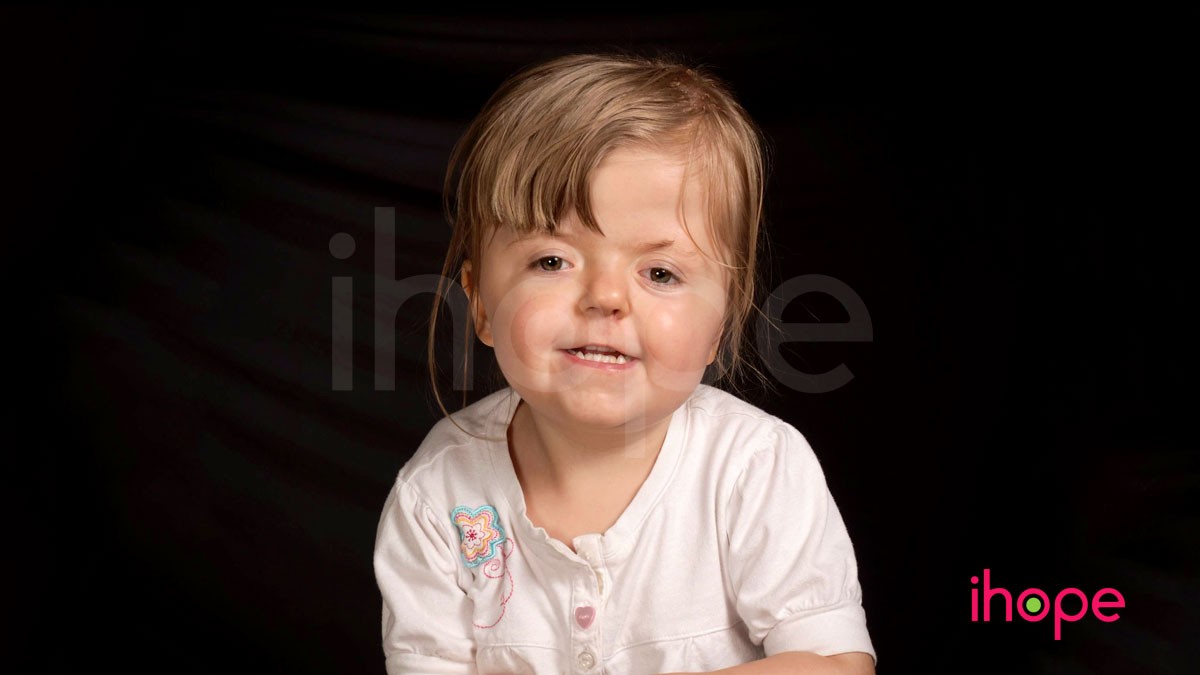
Hội chứng Apert là bệnh di truyền đặc trưng bởi các bất thường về xương. Một đặc điểm chính của hội chứng Apert là sự đóng sớm của xương hộp sọ (craniosynostosis) ngăn hộp sọ phát triển bình thường và ảnh hưởng đến hình dạng của đầu và mặt. Ngoài ra, một số ngón tay và ngón chân khác nhau dính lại với nhau.
Biểu hiện lâm sàng
Craniosynostosis gây ra nhiều đặc điểm trên khuôn mặt của hội chứng Apert. Sự hợp nhất sớm của các xương hộp sọ ngăn cản đầu phát triển bình thường, dẫn đến hình dạng trũng ở giữa mặt (chứng dị sản giữa mặt), mũi khoằm, trán nhăn nheo và vòm miệng bị hở (sứt môi). Ở những người mắc hội chứng Apert, hàm trên kém phát triển có thể dẫn đến các vấn đề về răng miệng, chẳng hạn như mất răng, men răng không đều và răng mọc chen chúc.
Nhiều người mắc hội chứng Apert có vấn đề về thị lực do các bất thường về mắt, có thể bao gồm mắt lồi, mắt mở rộng (cường tròng), góc ngoài của mắt hướng xuống dưới, mắt không nhìn vào cùng hướng (lé), và hốc mắt nông (lồi mắt). Một số người mắc hội chứng Apert bị mất thính lực hoặc nhiễm trùng tai tái phát do cấu trúc tai bị dị dạng.
Sự phát triển bất thường của các cấu trúc ở mặt và đầu cũng có thể gây tắc nghẽn một phần đường thở dẫn đến khó thở ở những người mắc hội chứng Apert. Craniosynostosis cũng ảnh hưởng đến sự phát triển của não, có thể làm gián đoạn sự phát triển trí tuệ. Khả năng nhận thức ở những người mắc hội chứng Apert từ bình thường đến khuyết tật trí tuệ nhẹ hoặc trung bình.
Những người mắc hội chứng Apert bị dính khớp ngón tay và ngón chân. Mức độ nghiêm trọng khác nhau, mặc dù bàn tay có xu hướng bị ảnh hưởng nghiêm trọng hơn bàn chân. Thông thường nhất, ba ngón trên mỗi bàn tay và bàn chân bị dính với nhau. Trong trường hợp nghiêm trọng nhất, tất cả các ngón tay và ngón chân đều bị dính lại. Hiếm khi những người bị hội chứng Apert có thêm ngón tay hoặc ngón chân. Một số người có bất thường ở xương khuỷu tay hoặc vai. Những vấn đề về xương này có thể hạn chế chuyển động và cản trở các hoạt động hàng ngày. Ở một số người, bất thường xảy ra ở cả hai bên của cơ thể, nhưng ở những người khác, chỉ một bên bị ảnh hưởng.
Các dấu hiệu và triệu chứng bổ sung của hội chứng Apert có thể bao gồm đổ mồ hôi nhiều bất thường (hyperhidrosis), da dầu bị mụn trứng cá nghiêm trọng hoặc thiếu lông mày.
Độ phổ biến
Hội chứng Apert ảnh hưởng đến khoảng 1/65.000 đến 1/88.000 trẻ sơ sinh. Mặc dù cha mẹ ở mọi lứa tuổi đều có thể có con mắc hội chứng Apert, nguy cơ sẽ tăng lên ở những ông bố lớn tuổi.
Nguyên nhân
Gen FGFR2 cung cấp hướng dẫn để tạo ra một protein được gọi là thụ thể 2 của yếu tố tăng trưởng nguyên bào sợi (FGFR2). Trong số nhiều chức năng, protein FGFR2 đóng một vai trò quan trọng trong sự phát triển trước khi sinh bằng cách truyền tín hiệu cho các tế bào chưa trưởng thành trở thành tế bào xương. Đột biến ở một phần cụ thể của gen FGFR2 làm thay đổi protein, dẫn đến gia tăng tín hiệu. Tín hiệu bất thường khiến tế bào trưởng thành quá nhanh và thúc đẩy quá trình hợp nhất sớm của xương trong hộp sọ, bàn tay và bàn chân.
Điều trị
Ở một đứa trẻ sơ sinh bình thường, hộp sọ được tạo thành từ một số “đĩa” được kết nối hờ, dần dần phát triển cùng nhau để tạo thành hộp sọ người lớn. Trong hội chứng Apert, các mảng này hợp nhất quá sớm làm hạn chế sự phát triển của não và gây tăng áp lực trong não khi nó lớn lên. Điều này được gọi là craniosynostosis. Phẫu thuật sớm để tách các mảng ra khỏi nhau giúp giảm áp lực. Phẫu thuật này thường diễn ra trong năm đầu tiên của cuộc đời, bác sĩ có thể thực hiện một số "tu sửa sọ não" để mang lại cho đứa trẻ một hình dáng bình thường hơn.
Khi hộp sọ phát triển, một phần ba giữa của khuôn mặt phát triển chậm hơn, dẫn đến sự tái tạo rõ rệt hơn theo thời gian. Một quy trình phẫu thuật được gọi là LeFort III có thể được sử dụng để điều chỉnh tình trạng này. Quy trình này thường được thực hiện sau giai đoạn phát triển quan trọng của tuổi thiếu niên và có thể lặp lại nếu cần. Quy trình LeFort liên quan đến việc tách da mặt xương từ giữa mắt đến hàm trên và kéo giãn vùng này ra bằng ghép xương sao cho căn chỉnh phù hợp. Nếu trán cũng không phát triển tốt, có thể sử dụng một thủ thuật gọi là "monoblock".
Trong vài năm gần đây, nhiều bác sĩ phẫu thuật đã thích "đánh lạc hướng" xương bằng cách sử dụng hệ thống RED hoặc các thiết bị đánh lạc hướng được đặt bên trong. Với quy trình này, thao tác vẫn như cũ nhưng giờ xương được kéo dần về phía trước thay vì di chuyển ngay lập tức trong quá trình phẫu thuật. Điều này dẫn đến sự hình thành xương mới theo thời gian.
Dạng di truyền
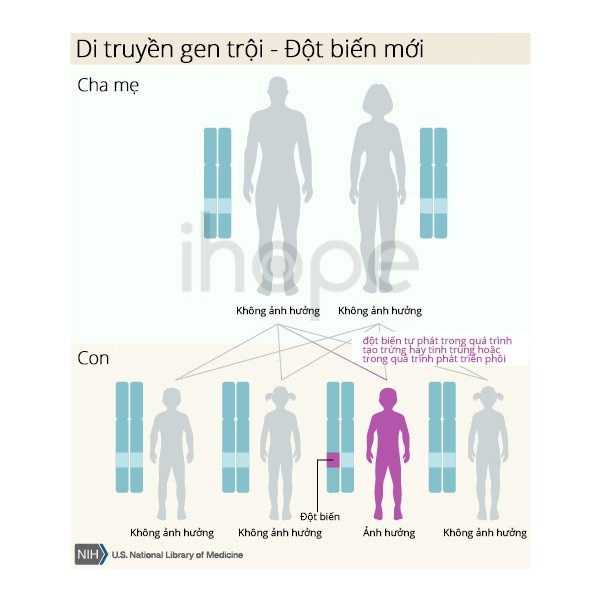
Hội chứng Apert được di truyền theo kiểu trội, nghĩa là chỉ cần đột biến trên một bản sao của gen chịu trách nhiệm trong mỗi tế bào là đủ để gây ra biểu hiện bệnh. Khi một người mang đột biến trội sinh con thì mỗi người con có 50% khả năng kế thừa đột biến đó.

Nguồn: U.S. National Library of Medicine
Trong một số trường hợp khác, đột biến xảy ra lần đầu tiên ở một người không có tiền sử gia đình mắc bệnh, được gọi là đột biến de novo.
Nguồn: U.S. National Library of Medicine
Phòng ngừa
Nếu một người mang hội chứng Apert, khi sinh con sẽ có 50% khả năng di truyền bệnh. Do đó, để bảo đảm 100% khả năng con không bị bệnh, cha mẹ có thể chọn phương pháp thụ tinh trong ống nghiệm IVF và sàng lọc phôi PGS/PGD.
Các tên gọi khác
- acrocephalosyndactyly
- acrocephalosyndactyly loại I
- Hội chứng Apert
- loại I acrocephalosyndactyly
References
- Genetic Testing Information. Acrocephalosyndactyly type I. Retrieved March 2, 2021 from https://www.ncbi.nlm.nih.gov/gtr/conditions/C0001193/
- Catalog of Genes and Diseases from OMIM. APERT SYNDROME. Retrieved March 2, 2021 from https://omim.org/entry/101200
- Genetic and Rare Diseases Information Center. Apert syndrome. Retrieved March 2, 2021 from https://rarediseases.info.nih.gov/diseases/5833/apert-syndrome
- U.S. National Library of Medicine. Apert syndrome. Retrieved January 29, 2021 from https://medlineplus.gov/genetics/condition/apert-syndrome/
- Children's Craniofacial Association. Apert syndrome. Retrieved January 29, 2021 from https://www.ccakids.org/assets/syndromebk_apert.pdf