Hội chứng Hajdu-Cheney là một bệnh di truyền hiếm gặp ảnh hưởng đến nhiều bộ phận của cơ thể, đặc biệt là xương. Mất mô xương từ bàn tay và bàn chân (acro-osteolysis) là một biểu hiện đặc trưng. Các ngón tay, ngón chân ngắn và rộng, ngắn dần theo thời gian do xương ở các đầu bị gãy liên tục.
Biểu hiện lâm sàng
Các bất thường về xương trên toàn cơ thể thường gặp trong hội chứng Hajdu-Cheney. Người bệnh phát triển chứng loãng xương, làm cho xương giòn và dễ bị gãy, nhiều người bị gãy xương cột sống (đốt sống). Một số khác có cột sống cong bất thường (vẹo cột sống hoặc gù cột sống). Hội chứng Hajdu-Cheney cũng ảnh hưởng đến hình dạng và sức mạnh của xương dài ở tay và chân, tầm vóc thấp.
Hội chứng Hajdu-Cheney cũng gây ra các bất thường của xương sọ, bao gồm cả xương khuôn mặt. Hình dạng của hộp sọ thường được mô tả là dolichocephalic thuôn dài từ sau ra trước. Ở nhiều người bệnh, xương ở phía sau hộp sọ phình ra, gây một vết sưng gọi là chẩm nổi rõ. Các dị biệt trên khuôn mặt như mắt có khoảng cách rộng và xếch xuống, lông mày mọc ở giữa, tai cụp xuống, phần giữa mặt có vẻ trũng xuống và khoảng trống lớn giữa mũi và môi trên. Một số trẻ sinh ra với khe hở ở vòm miệng được gọi là sứt môi hoặc vòm miệng cao. Ở người lớn bị ảnh hưởng, các đặc điểm trên khuôn mặt thường được mô tả là "thô".
Các đặc điểm khác của hội chứng Hajdu-Cheney bao gồm các bất thường về khớp, đặc biệt là phạm vi cử động khớp lớn bất thường (hypermobility), vấn đề nha khoa, mất thính lực, giọng nói trầm khàn, lông thừa trên cơ thể, nhiễm trùng tái phát trong thời thơ ấu, dị tật tim và các bất thường về thận như thận đa nang. Một số người bị chậm phát triển trong thời thơ ấu, nhưng thường nhẹ.
Các biến chứng nghiêm trọng nhất của hội chứng Hajdu-Cheney xảy ra ở khoảng một nửa số người bị ảnh hưởng là chứng platybasia và basilar invagination. Platybasia là tình trạng phẳng đáy hộp sọ do xương sọ mỏng và mềm. Basilar invaginationxảy ra khi xương mềm cho phép một phần của cột sống nhô ra bất thường qua lỗ ở đáy hộp sọ, đẩy vào các phần dưới của não. Những bất thường này có thể dẫn đến các vấn đề thần kinh nghiêm trọng, bao gồm đau đầu, bất thường về thị lực và thăng bằng, tích tụ chất lỏng trong não (não úng thủy), thở bất thường và đột tử.
Các dấu hiệu và triệu chứng của hội chứng Hajdu-Cheney rất khác nhau giữa các cá nhân bị ảnh hưởng, ngay cả giữa các thành viên trong cùng một gia đình. Nhiều đặc điểm của bệnh như chứng tiêu xương và một số đặc điểm đặc trưng trên khuôn mặt không xuất hiện khi mới sinh nhưng trở nên rõ ràng trong thời thơ ấu hoặc sau đó. Nguy cơ phát triển platybasia và basilar invagination cũng tăng lên theo thời gian.
Các đặc điểm của hội chứng Hajdu-Cheney trùng lặp đáng kể với các đặc điểm của một tình trạng gọi là hội chứng thận đa nang hình sợi huyết thanh ngoằn ngoèo (SFPKS). Mặc dù chúng từng được coi là những rối loạn riêng biệt, các nhà nghiên cứu đã phát hiện ra rằng hai tình trạng này có liên quan đến các đột biến trên cùng một gen. Dựa trên những điểm tương đồng này, nhiều nhà nghiên cứu hiện coi hội chứng Hajdu-Cheney và SFPKS là những biến thể của cùng một tình trạng.
Độ phổ biến
Hội chứng Hajdu-Cheney hiếm gặp, mức độ phổ biến vẫn chưa được thống kê. Chưa đến 100 trường hợp được ghi nhận trong các tài liệu y tế.
Nguyên nhân
Gen NOTCH2 cung cấp hướng dẫn để tạo ra một thụ thể được gọi là Notch2. Các thụ thể kết hợp với protein ligand, khớp với nhau giống như chìa và ổ khóa. Khi một ligand liên kết với thụ thể Notch2, nó sẽ kích hoạt các tín hiệu quan trọng cho sự phát triển bình thường và chức năng của nhiều loại tế bào khác nhau. Các nghiên cứu cho thấy rằng tín hiệu thông qua thụ thể Notch2 rất quan trọng cho sự phát triển ban đầu của xương và sau này cho quá trình tái tạo xương, một quá trình bình thường trong đó xương cũ bị loại bỏ và xương mới được tạo ra để thay thế. Tín hiệu Notch2 dường như cũng liên quan đến sự phát triển của tim, thận, răng và các bộ phận khác của cơ thể.
Các đột biến ở một khu vực cụ thể gần cuối gen NOTCH2 có liên quan đến hội chứng Hajdu-Cheney. Những đột biến này dẫn đến một phiên bản của thụ thể Notch2 không thể bị phá vỡ như bình thường. Kết quả là thụ thể tiếp tục hoạt động ngay cả khi có tín hiệu dừng lại. Các nhà nghiên cứu chưa rõ tín hiệu Notch2 quá mức có liên quan như thế nào đến nhiều biểu hiện đa dạng của hội chứng Hajdu-Cheney. Họ nghi ngờ rằng các vấn đề về xương bao gồm chứng tiêu xương, loãng xương và những dị biệt trên khuôn mặt, có khả năng do quá trình tái tạo và phát triển xương bất thường. Tín hiệu dư thừa thông qua thụ thể Notch2 hoạt động quá mức có thể làm tăng quá trình loại bỏ xương cũ, giảm sự hình thành xương mới hoặc cả hai. Chưa rõ bằng cách nào mà thụ thể hoạt động quá mức góp phần vào các dấu hiệu và triệu chứng khác.
Điều trị
Hội chứng Hajdu-Cheney biểu hiện khác nhau ở mỗi trường hợp, với những khiếm khuyết ở mặt hay tay chân, có thể phẫu thuật để chỉnh sửa.
Dạng di truyền
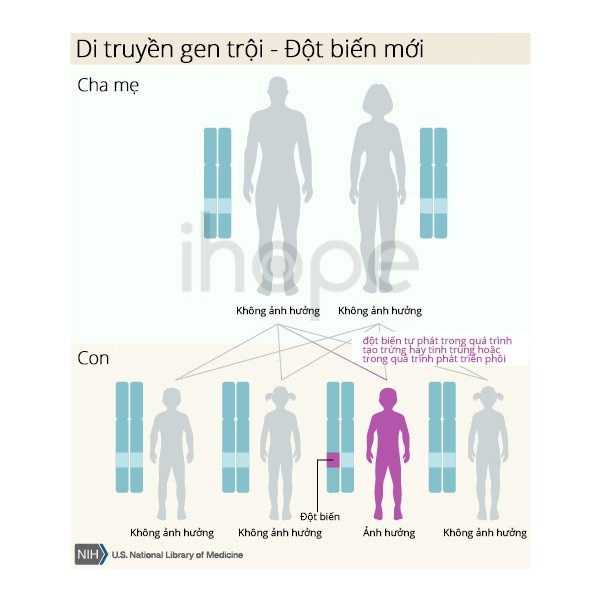
Bệnh được di truyền theo kiểu trội trên nhiễm thường, có nghĩa là một bản sao của gen NOTCH2 bị thay đổi trong mỗi tế bào là đủ để gây bệnh. Hầu hết các trường hợp là do đột biến gen mới và xảy ra ở những người không có tiền sử mắc chứng rối loạn này trong gia đình họ. Ít phổ biến hơn, người bệnh thừa hưởng đột biến từ cha hoặc mẹ bị ảnh hưởng.
Nguồn: U.S. National Library of Medicine
Phòng ngừa
Bệnh chủ yếu do đột biến mới phát sinh nên rất khó để phòng ngừa. Trường hợp bố hoặc mẹ mang bệnh, khi sinh con có 50% khả năng thừa hưởng bệnh, do đó cần làm thụ tinh nhân tạo IVF kết hợp sàng lọc phôi PGS/PGD để bảo đảm sinh con khỏe mạnh.
Các tên gọi khác
- acroosteolysis trội
- acroosteolysis với loãng xương và những thay đổi trong hộp sọ và xương hàm dưới
- loạn sản khớp-răng-xương
- arthrodentoosteodysplasia
- Hội chứng Cheney
- loạn sản xương sọ với tiêu xương acro
- loạn sản xương gia đình
- loạn sản xương di truyền với tiêu xương acro-osteolysis
- HJCYS
- hội chứng thận đa nang hình sợi huyết thanh ngoằn ngoèo
- SFPKS
References
- Genetic Testing Information. Hajdu-Cheney syndrome. Retrieved March 8, 2021 from https://www.ncbi.nlm.nih.gov/gtr/conditions/C0917715/
- Catalog of Genes and Diseases from OMIM. HAJDU-CHENEY SYNDROME. Retrieved March 8, 2021 from https://omim.org/entry/102500/
- Genetic and Rare Diseases Information Center. Acroosteolysis dominant type. Retrieved March 8, 2021 from https://rarediseases.info.nih.gov/diseases/508/acroosteolysis-dominant-type
- U.S. National Library of Medicine. Hajdu-Cheney syndrome. Retrieved March 8, 2021 from https://medlineplus.gov/genetics/condition/hajdu-cheney-syndrome/