
Hội chứng McKusick-Kaufman (McKusick-Kaufman syndrome) là bệnh di truyền hiếm gặp. Bệnh ảnh hưởng đến quá trình hình thành chi, tim và hệ thống sinh sản.
Biểu hiện lâm sàng
Bệnh có 3 đặc điểm bao gồm tật thừa ngón (ngón tay hoặc ngón chân), dị tật tim và bộ phận sinh dục bất thường. Bé gái bị ứ dịch vùng âm đạo (hydrometrocolpos) do tắc nghẽn âm đạo trước sinh. Trẻ có một khối u nang lớn chứa chất lỏng tích tụ trong khung chậu. Bé trai có lỗ tiểu thấp, dương vật cong xuống và tinh hoàn chưa xuống bìu.
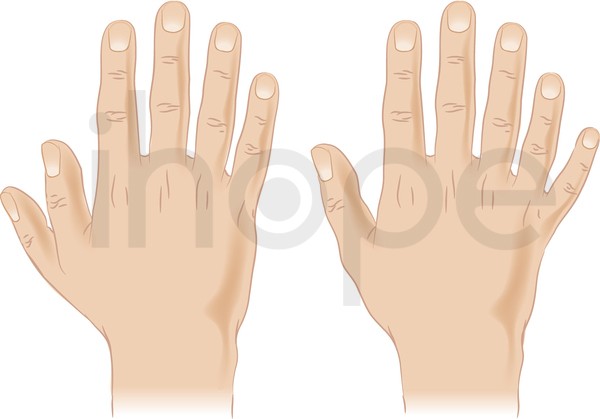
Ảnh: Dị tật thừa ngón tay
Dị tật thừa ngón tay (polydactyly) xuất hiện như một phần của tập hợp các bất thường. Polydactyly có thể tồn tại độc lập, trong trường hợp này nó được di truyền kiểu trội trên nhiễm sắc thể thường.
Nguồn: Darryl Leja, NHGRI[/caption]
Độ phổ biến
Hội chứng McKusick-Kaufman là bệnh di truyền hiếm gặp được phát hiện đầu tiên tại quần thể Old Order Amish. Tỷ lệ mắc bệnh tại quần thể này khoảng 1/10.000 người. Những chủng tộc người khác chưa thống kê được số người mắc bệnh.
Nguyên nhân
Đột biến gen MKKS gây ra hội chứng McKusick-Kaufman. Gen MKKS cung cấp hướng dẫn tạo ra protein nắm vai trò quan trọng trong quá trình hình thành các chi, tim và hệ thống sinh sản. Protein này có cấu trúc như chaperonin, nó giúp protein khác gấp cuộn. Tuy nhiên, người ta cho rằng protein MKKS có thể tham gia vào quá trình vận chuyển các protein khác trong tế bào.
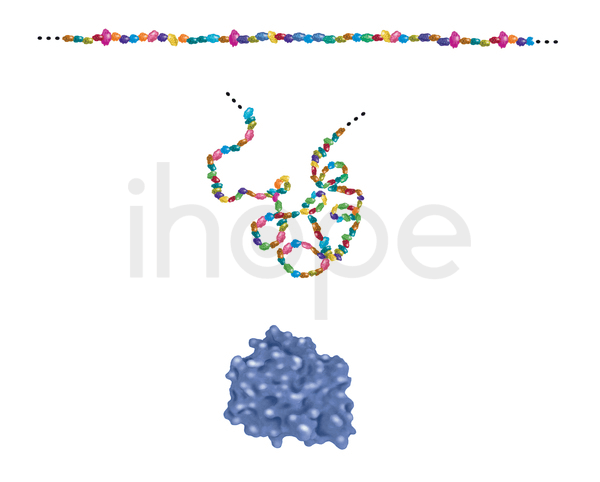
Protein cấu tạo từ các axit amin và gấp cuộn để thực hiện chức năng của nó.
Nguồn: National Institute of General Medical Sciences/National Institutes of Health
Đột biến gen làm thay đổi cấu trúc của protein MKKS. Protein đột biến làm gián đoạn quá trình phát triển của một số bộ phận trong cơ thể trước khi sinh. Tuy nhiên, người ta vẫn chưa hiểu rõ vì sao đột biến MKKS gây ra các dấu hiệu và triệu chứng của hội chứng McKusick-Kaufman.
Chẩn đoán
Người bệnh được chẩn đoán bệnh thông qua các biểu hiện lâm sàng và tiền sử bệnh trong gia đình. Một số xét nghiệm chuyên biệt như:
- Xét nghiệm hình ảnh (chụp MRI, chụp CT, siêu âm) vùng bụng và xương các chi
- Nội soi bàng quang, âm đạo.
- Xét nghiệm di truyền tìm đột biến gen MKKS
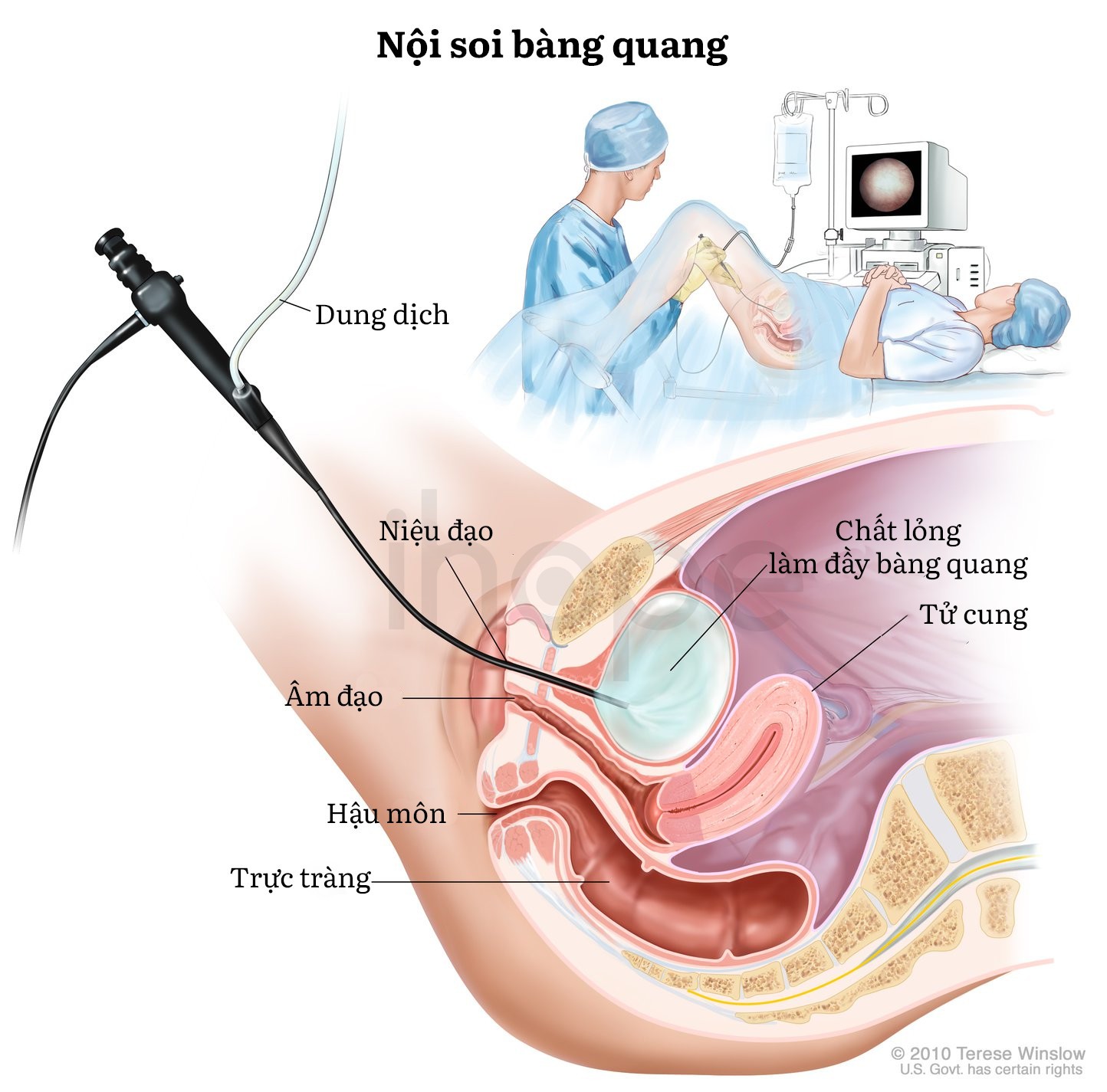
Nguồn: National Institute of Diabetes Digestive and Kidney Diseases
Biểu hiện lâm sàng của hội chứng McKusick-Kaufman tương tự với hội chứng Bardet-Biedl. Tuy nhiên, hội chứng Bardet-Biedl có một số đặc điểm không thấy trong hội chứng McKusick-Kaufman, bao gồm giảm thị lực, chậm phát triển, béo phì và suy thận. Những đặc điểm này chưa biểu hiện rõ ràng khi mới sinh, do đó khó phân biệt hai hội chứng này ở trẻ sơ sinh và trẻ nhỏ.
Điều trị
Hiện nay chưa có phương pháp điều trị hoàn toàn hội chứng McKusick-Kaufman. Một số liệu pháp giúp khắc phục triệu chứng bệnh như:
- Phẫu thuật dẫn lưu hút dịch lỏng ứ trong vùng âm đạo ra ngoài. Sau phẫu thuật, bệnh nhân cần được theo dõi tránh biến chứng nhiễm trùng đường tiết niệu tái phát và tái hẹp lỗ âm đạo.
- Phẫu thuật điều chỉnh các dị tật bộ phận sinh dục hoặc xương chi.
Dạng di truyền
Bệnh di truyền theo kiểu lặn trên nhiễm sắc thể thường. Do đó, bệnh chỉ biểu hiện khi có cả hai bản sao của gen đột biến trong mỗi tế bào. Bệnh nhân mắc bệnh lặn trên nhiễm sắc thể thường sẽ có bố và mẹ mang một bản sao của gen đột biến, nhưng bố mẹ ít khi biểu hiện triệu chứng bệnh.

Nguồn: U.S. National Library of Medicine
Phòng ngừa
Hội chứng McKusick-Kaufman di truyền lặn trên nhiễm sắc thể thường, cha mẹ mang đột biến dị hợp nên gần như không có biểu hiện bệnh, do đó rất khó phát hiện cho đến khi sinh con. Để chủ động phòng ngừa, cha mẹ nên làm xét nghiệm sàng lọc gen lặn để chủ động cho tương lai của con.
Các thành viên trong gia đình nên khám sức khỏe và tầm soát bệnh định kỳ nếu có thành viên mắc bệnh. Các cặp vợ chồng trước khi mang thai cần tư vấn và xét nghiệm di truyền đảm bảo sinh con khỏe mạnh.
Các tên gọi khác
- HMCS
- Hydrometrocolpos, postaxial polydactyly, and congenital heart malformation
- Kaufman-McKusick syndrome
- MKS
References
- Genetic Testing Information. McKusick-Kaufman syndrome. Retrieved October 31, 2022 from https://www.ncbi.nlm.nih.gov/gtr/conditions/C0948368/
- Genetic and Rare Diseases Information Center. McKusick-Kaufman syndrome. Retrieved October 31, 2022 from https://rarediseases.info.nih.gov/diseases/3427/mckusick-kaufman-syndrome/
- Catalog of Genes and Diseases from OMIM. MCKUSICK-KAUFMAN SYNDROME. Retrieved October 31, 2022 from https://omim.org/entry/236700
- U.S National Library of Medicine. McKusick-Kaufman syndrome. Retrieved October 31, 2022 from https://medlineplus.gov/genetics/condition/mckusick-kaufman-syndrome/
- Orphanet. McKusick-Kaufman syndrome. Retrieved October 31, 2022 from https://www.orpha.net/consor/cgi-bin/OC_Exp.php?Lng=GB&Expert=2473
- MalaCards. Mckusick-Kaufman Syndrome (MKKS). Retrieved October 31, 2022 from https://www.malacards.org/card/mckusick_kaufman_syndrome
- David A, Bitoun P, Lacombe D, Lambert JC, Nivelon A, Vigneron J, Verloes A. Hydrometrocolpos and polydactyly: a common neonatal presentation of Bardet-Biedl and McKusick-Kaufman syndromes. J Med Genet. 1999 Aug;36(8):599-603. doi 10.1136/jmg.36.8.599.
- David A, Bitoun P, Lacombe D, Lambert JC, Nivelon A, Vigneron J, Verloes A. Hydrometrocolpos and polydactyly: a common neonatal presentation of Bardet-Biedl and McKusick-Kaufman syndromes. J Med Genet. 1999 Aug;36(8):599-603.
- Slavotinek AM, Biesecker LG. Phenotypic overlap of McKusick-Kaufman syndrome with bardet-biedl syndrome: a literature review. Am J Med Genet. 2000 Nov 27;95(3):208-15.
- Slavotinek AM. McKusick-Kaufman Syndrome. 2002 Sep 10 [updated 2020 Dec 3]. In: Adam MP, Everman DB, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2022. Available from http://www.ncbi.nlm.nih.gov/books/NBK1502/
- Stone DL, Slavotinek A, Bouffard GG, Banerjee-Basu S, Baxevanis AD, Barr M, Biesecker LG. Mutation of a gene encoding a putative chaperonin causes McKusick-Kaufman syndrome. Nat Genet. 2000 May;25(1):79-82.





