Hội chứng Bardet-Biedl là một bệnh di truyền ảnh hưởng đến nhiều bộ phận của cơ thể. Các biểu hiện đặc trưng bao gồm suy giảm thị lực, béo phì, thừa ngón và sinh sản kém.
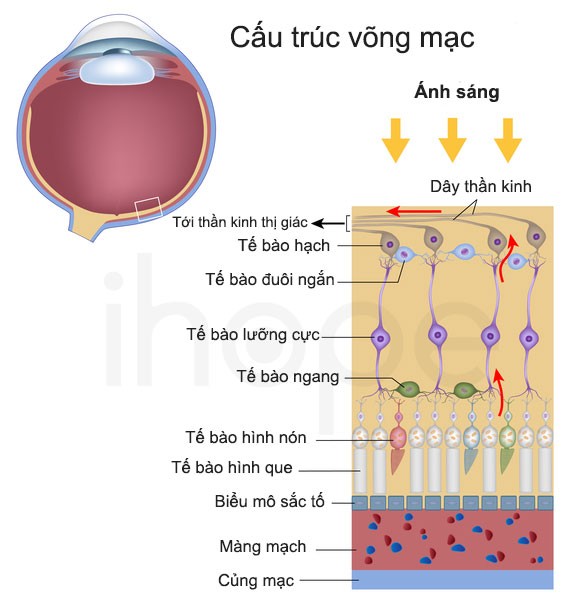
Ảnh: Cấu trúc võng mạc
Nguồn: U.S. National Library of Medicine
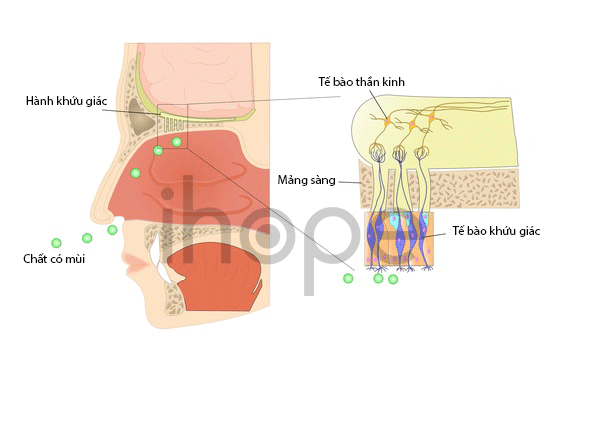
Ảnh: Hệ thống khứu giác
Nguồn: U.S. National Library of Medicine

Ảnh: Ngón tay ngắn bất thường
Nguồn: U.S. National Library of Medicine
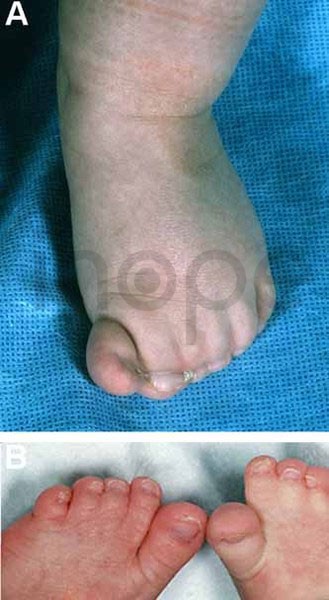
Ảnh: Tật dính ngón
Nguồn: U.S. National Library of Medicine
Biểu hiện lâm sàng
Mất thị lực là một trong những đặc điểm chính của hội chứng Bardet-Biedl khi các mô cảm nhận ánh sáng nằm phía sau mắt (võng mạc ) dần suy giảm. Giảm thị lực biểu hiện rõ ràng ban đêm khi trẻ còn nhỏ, sau đó phát triển các điểm mù ở vùng ngoại biên. Theo thời gian, những điểm mù này mở rộng và hợp nhất tạo thành tầm nhìn hình ống. Hầu hết những người mắc hội chứng Bardet-Biedl thường có thị lực kém và có thể bị mù ở tuổi vị thành niên hoặc giai đoạn đầu trưởng thành.
Béo phì là một triệu chứng đặc trưng khác của hội chứng Bardet-Biedl. Tăng cân bất thường thường bắt đầu từ thời thơ ấu và tiếp tục kéo dài suốt đời. Các biến chứng của béo phì có thể bao gồm bệnh tiểu đường tuýp 2, tăng huyết áp và cholesterol cao bất thường (tăng cholesterol máu).
Các dấu hiệu và triệu chứng khác bao gồm thừa ngón tay hoặc ngón chân (nhiều ngón), thiểu năng trí tuệ hoặc học tập kém và bất thường cơ quan sinh dục. Hầu hết nam giới mắc bệnh đều giảm sản xuất hormone sinh dục (thiểu năng sinh dục) có thể dẫn đến vô sinh nam. Nhiều người mắc hội chứng Bardet-Biedl cũng có bất thường về thận đến mức nghiêm trọng hoặc đe dọa tính mạng.
Người bệnh có thể chậm nói, chậm phát triển kỹ năng vận động như đi đứng, các vấn đề hành vi như vụng về hoặc phối hợp kém, khuôn mặt dị biệt, bất thường răng miệng, ngón tay hoặc ngón chân ngắn hoặc dính vào nhau . Mất khứu giác một phần hoặc hoàn toàn cũng đã được báo cáo ở một số người mắc hội chứng Bardet-Biedl. Ngoài ra, bệnh còn ảnh hưởng đến tim, gan và hệ tiêu hóa.
Độ phổ biến
Ở hầu hết khu vực Bắc Mỹ và Châu Âu, hội chứng Bardet-Biedl có tỷ lệ hiện mắc từ 1/140.000 đến 1/160.000 trẻ sơ sinh. Bệnh phổ biến hơn ở đảo Newfoundland (ngoài khơi bờ biển phía đông Canada), ước tính 1/17.000 trẻ. Đối với người Bedouin ở Kuwait (quốc gia ở khu vực Trung Đông), hội chứng ảnh hưởng đến khoảng 1/13.500 trẻ.
Nguyên nhân
Hội chứng Bardet-Biedl có thể do đột biến ít nhất 14 gen khác nhau (thường gọi là gen BBS). Những gen này đóng những vai trò quan trọng trong cấu trúc tế bào được gọi là lông mao. Lông mao là những sợi cực nhỏ, nhô ra khỏi bề mặt của nhiều loại tế bào. Chúng tham gia vào chuyển động của tế bào và nhiều đường tín hiệu hóa học khác nhau. Lông mao cũng cần thiết cho nhận thức của các đầu vào giác quan (như thị giác, thính giác và khứu giác). Protein được tạo ra từ các gen BBS có liên quan đến cơ chế duy trì và hoạt động của lông mao.
Các đột biến gen BBS gây ra nhiều vấn đề với cấu trúc và chức năng của lông mao. Những khiếm khuyết này có thể làm gián đoạn các đường tín hiệu hóa học quan trọng trong quá trình phát triển, dẫn đến những bất thường về nhận thức cảm giác. Người ta tin rằng lông mao bị khiếm khuyết là nguyên nhân gây ra hầu hết các triệu chứng của hội chứng Bardet-Biedl.
Khoảng 25% trường hợp mắc hội chứng Bardet-Biedl do đột biến gen BBS1. 20% trường hợp khác do đột biến gen BBS10. Các đột biến gen BBS khác chỉ chiếm một tỉ lệ nhỏ trong tất cả các trường hợp mắc bệnh. Khoảng 25% mắc hội chứng Bardet-Biedl chưa rõ nguyên nhân.
Ở những bệnh nhân mang đột biến ở một trong các gen BBS, đột biến tại các gen khác có thể liên quan đến sự biến đổi trong các dấu hiệu và triệu chứng của hội chứng Bardet-Biedl. Nghiên cứu cho thấy những gen đột biến bổ sung có thể là gen BBS đã biết hoặc các gen khác. Tuy nhiên, hiện tượng này dường như không phổ biến và chưa nhất quán.
Chẩn đoán
Hội chứng Bardet-Biedl được chẩn đoán dựa trên các biểu hiện trưng như các vấn đề về thị giác do loạn dưỡng võng mạc, béo phì, thừa ngón. Bởi vì dựa trên các phát hiện lâm sàng, một số bệnh nhân có thể không được chẩn đoán chính xác trong thời gian dài.
Khó khăn trong quá trình chẩn đoán phát sinh khi trẻ có biểu hiện khó học tập và vấn đề cân nặng nhưng lại không có bất kỳ dị tật bẩm sinh nào. Đối với những trường hợp này, chẩn đoán có thể vẫn chưa chắc chắn cho đến khi các vấn đề thị lực bắt đầu biểu hiện. Chẩn đoán bệnh võng mạc bằng cách đo các phản ứng điện của các loại tế bào khác nhau trong võng mạc (electroretinogram - ERG).
Xét nghiệm di truyền có thể giúp xác định chẩn đoán ở một số bệnh nhân (ví dụ: những người có đột biến gen BBS1 và BBS10).
Điều trị
Mục tiêu điều trị chính cho bệnh nhân Bardet-Biedl là tập trung vào các triệu chứng cụ thể.
Các vấn đề về mắt
Triệu chứng đầu tiên khởi phát thường là quáng gà phổ biến ở độ tuổi 8-9. Thiếu vitamin A có thể làm trầm trọng thêm các vấn đề thị giác, vitamin bổ sung và khoáng chất phù hợp với lứa tuổi có thể hỗ trợ chức năng mắt. Mặc dù hiện tại chưa có liệu pháp chữa khỏi chứng loạn dưỡng võng mạc, người bệnh vẫn cần phải theo dõi thường xuyên. Bác sĩ có thể điều chỉnh các tật khúc xạ (ví dụ: cận thị hoặc viễn thị) và thị lực kém.
Béo phì
Béo phì thường biểu hiện ở độ tuổi từ hai đến ba tuổi. Lối sống năng động kết hợp với thể dục thể thao có thể cải thiện đáng kể. Chế độ ăn uống tốt có thể ngăn ngừa các vấn đề liên quan đến cân nặng trong cuộc sống sau này. Người bệnh cần tham khảo ý kiến của bác sĩ và chuyên gia dinh dưỡng để lập chế độ ăn uống đầy đủ và ngăn ngừa tăng cân quá mức. Các vấn đề cholesterol cao và tiểu đường sẽ được điều trị theo các phương pháp thông dụng hiện nay.
Phẫu thuật
Một số bất thường thể chất liên quan đến hội chứng Bardet-Biedl có thể được điều chỉnh bằng phẫu thuật, bao gồm thừa ngón, bất thường hệ sinh dục và dị tật tim bẩm sinh. Nếu bệnh thận nghiêm trọng, có thể thực hiện ghép thận cho bệnh nhân. Phẫu thuật cần được đặc biệt quan tâm đối với bệnh nhân Bardet-Biedl. Gây mê toàn thân đòi hỏi một loạt các bước phối hợp dựa trên giải phẫu của đường thở. Một số bệnh nhân còn có bất thường trong đường thở, dẫn đến khó giữ đường thở trong khi gây mê toàn thân.
Vấn đề hormone
Những bệnh nhân có nồng độ hormone thấp có thể được kê đơn thuốc bổ sung bởi bác sĩ chuyên khoa nội tiết.
Dạng di truyền
Hội chứng Bardet-Biedl thường được di truyền theo kiểu lặn trên nhiễm sắc thể thường, nghĩa là cả hai bản sao của gen BBS trong mỗi tế bào đều có đột biến. Bố mẹ của một người mắc bệnh thường mang một bản sao của gen đột biến, nhưng họ thường không biểu hiện các dấu hiệu và triệu chứng của bệnh.

Nguồn: U.S. National Library of Medicine
Phòng ngừa
Hội chứng Bardet-Biedl thường được di truyền theo cơ chế gen lặn rất khó phát hiện ở những người thể mang vì gần như không có biểu hiện rõ ràng, đến khi có con mới biết được thì đã quá muộn. Do đó, các cặp vợ chồng trước khi mang thai cần làm xét nghiệm gen lặn để sàng lọc căn bệnh này, đảm bảo sinh con khỏe mạnh và lành lặn.
Các tên gọi khác
- BBS
References
- Genetic Testing Information. Bardet-Biedl syndrome. Retrieved September 24, 2021 from https://www.ncbi.nlm.nih.gov/gtr/conditions/C0752166/
- Catalog of Genes and Diseases from OMIM. BARDET-BIEDL SYNDROME 1; BBS1. Retrieved September 19, 2021 from https://omim.org/entry/209900
- Genetic and Rare Diseases Information Center. Bardet-Biedl syndrome. Retrieved September 24, 2021 from https://rarediseases.info.nih.gov/diseases/6866/bardet-biedl-syndrome
- U.S. National Library of Medicine. Bardet-Biedl syndrome. Retrieved September 19, 2021 from https://medlineplus.gov/genetics/condition/bardet-biedl-syndrome/
- National Organization for Rare Disorders (NORD). Bardet-Biedl Syndrome. Retrieved September 19, 2021 from https://rarediseases.org/rare-diseases/bardet-biedl-syndrome/
- Bardet Biedl Syndrome Foundation. What is BBS? Retrieved September 19, 2021 from https://www.bardetbiedl.org/what-is-bbs
- National Center for Advancing Translational Sciences. Bardet-Biedl syndrome. Retrieved September 19, 2021 from https://rarediseases.info.nih.gov/diseases/6866/bardet-biedl-syndrome
- Foundation Fighting Blindness. Bardet-Biedl syndrome (BBS). Retrieved September 19, 2021 from https://www.fightingblindness.org/diseases/bardet-biedl-syndrome-bbs