Hội chứng Joubert là bệnh di truyền hiếm gặp ở trẻ sơ sinh do bộ não phát triển bất thường. Hầu hết trẻ mắc hội chứng Joubert chậm phát triển, giảm chức năng gan hoặc thận. Các thành viên trong gia đình cùng mắc bệnh có thể biểu hiện triệu chứng lâm sàng khác nhau.
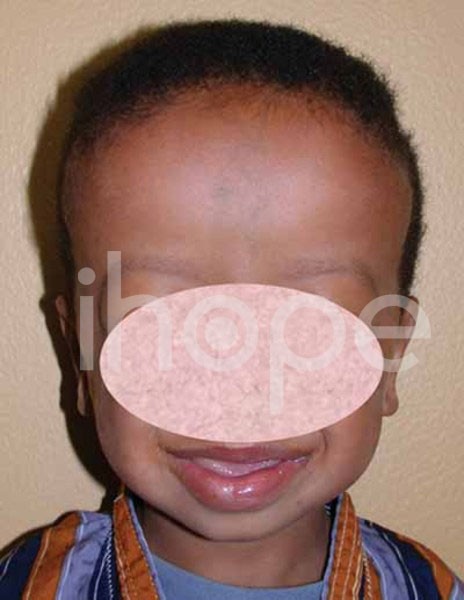
Ảnh: Trán rộng
Nguồn: National Library of Medicine
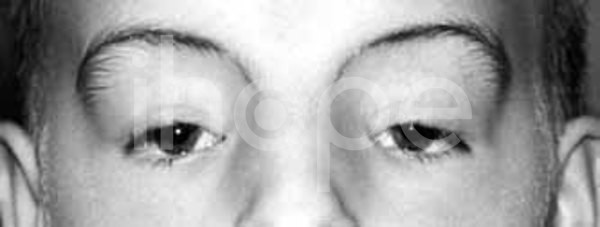
Ảnh: Lông mày cong
Nguồn: U.S National Library of Medicine
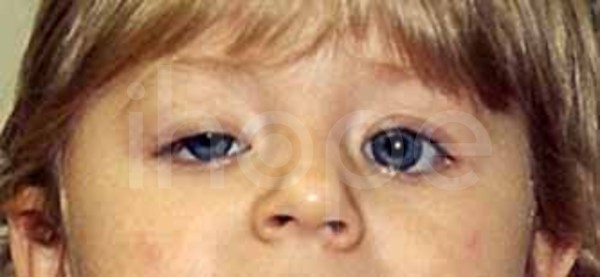
Ảnh: Mí mắt sụp xuống
Nguồn: U.S National Library of Medicine
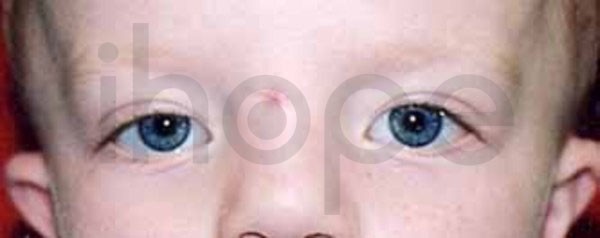
Ảnh: Khoảng cách giữ hai mắt rộng
Nguồn: Elements of Morphology, National Human Genome Research Institute
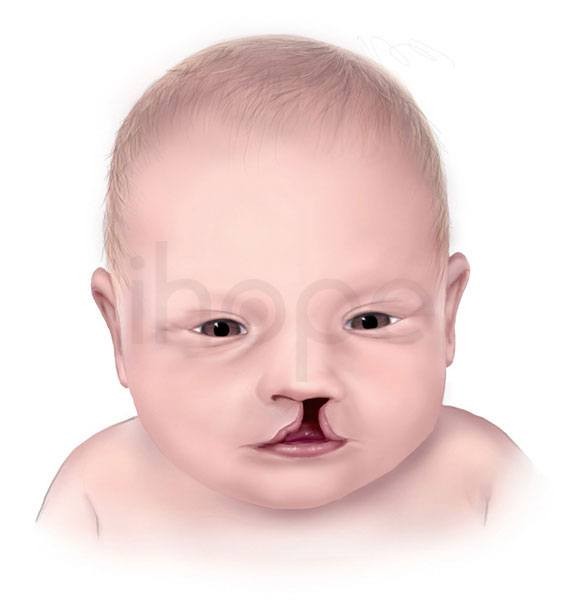
Ảnh: Sứt môi
Nguồn: Centers for Disease Control and Prevention
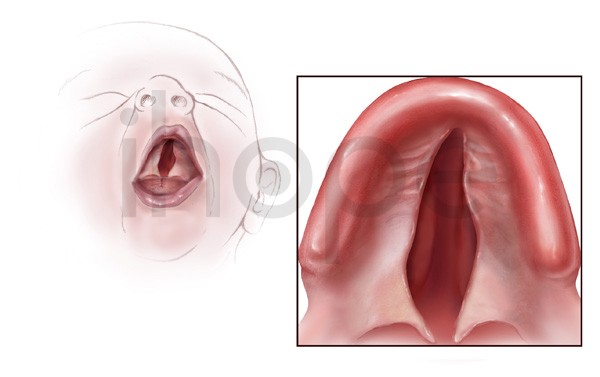
Ảnh: Hở hàm ếch
Nguồn: U.S National Library of Medicine
Biểu hiện lâm sàng
Hầu hết trẻ sơ sinh mắc hội chứng Joubert bị yếu cơ, mất điều hòa cử động, chậm phát triển và thiểu năng trí tuệ từ mức nhẹ đến nặng.
Một số đặc điểm khác của bệnh như:
- Hơi thở nhanh bất thường hoặc chậm (ngưng thở)
- Chuyển động mắt bất thường, ví dụ khó di chuyển mắt từ bên này sang bên kia
- Trán rộng
- Lông mày cong
- Mí mắt sụp
- Khoảng cách hai mắt rộng
- Tai thấp
- Sứt môi hoặc hở hàm ếch
- Thừa ngón tay hoặc ngón chân
Biến chứng
Trẻ mắc bệnh có thể phát triển một số biến chứng khác, bao gồm:
- Loạn dưỡng võng mạc gây mất thị lực
- Tật khuyết của mắt - một lỗ hổng trong cấu trúc của mắt có thể ảnh hưởng đến mí mắt, mống mắt, võng mạc, hoặc thần kinh thị giác của một hoặc cả hai mắt
- Tổn thương gan, thận (thận đa nang hoặc suy thận)
- Bất thường xương (tật nhiều ngón)
- Các vấn đề nội tiết
Độ phổ biến
Tỷ lệ mắc hội chứng Joubert khoảng 1/80.000 - 1/100.000 trẻ sơ sinh. Tuy nhiên,con số này có thể thấp hơn thực tế vì nhiều trường hợp chưa được chẩn đoán. Các đột biến gen gây bệnh xuất hiện phổ biến trong một số chủng tộc như người Do Thái Ashkenazi, người Canada gốc Pháp và người Hutterite.
Nguyên nhân
Hơn 30 gen đã được xác định gây ra hội chứng Joubert. Protein được tạo ra từ các gen này chưa rõ chức năng. Người ta cho rằng chúng tham gia vào cấu trúc tế bào được gọi là lông mao sơ cấp. Lông mao là những cấu trúc sợi cực nhỏ nằm trên bề mặt tế bào, chúng đóng vai trò cảm biến môi trường vật chất và truyền tín hiệu hóa học. Chúng có trong tế bào thần kinh, thận và gan. Lông mao cũng cần thiết để nhận tín hiệu đầu vào của các giác quan như thị giác, thính giác và khứu giác.
Các đột biến gen liên quan đến hội chứng Joubert gây ra những vấn đề với cấu trúc và chức năng của lông mao sơ cấp. Khiếm khuyết trong các cấu trúc tế bào này có thể phá vỡ đường dẫn truyền tín hiệu hóa học quan trọng trong quá trình phát triển. Người ra vẫn chưa hiểu rõ cơ chế các đột biến dẫn đến những đặc điểm bất thường của bệnh.
Khoảng 60-90% trường hợp mắc bệnh do đột biến các gen liên quan, bao gồm:
- Đột biến gen AHI1 gây ra khoảng 7-10% trường hợp mắc bệnh. Bệnh nhân thường suy giảm thị lực do chứng loạn dưỡng võng mạc.
- Đột biến gen NPHP1 (hay gen JBTS4) gây ra khoảng 1-2% trường hợp mắc bệnh. Bệnh thường biến chứng gây suy thận.
- Đột biến gen CEP290
- Đột biến gen OFD1, CPLANE1, CC2D2A, INP5E, KIAA0586, MKS1, RPGRIP1L TCTN2, TMEM67 và TMEM216 cũng gây ra hội chứng Joubert nhưng ít phổ biến.
Những trường hợp còn lại chưa rõ nguyên nhân di truyền.
Chẩn đoán
Người bệnh được khám sức khỏe, thăm hỏi về tiền sử bệnh của gia đình. Trường hợp nghi ngờ mắc hội chứng Joubert, bệnh nhân có thể thực hiện xét nghiệm hình ảnh. Đặc điểm nổi bật của hội chứng Joubert là các bất thường ở não, chúng được gọi là dấu hiệu molar tooth trên xét nghiệm hình ảnh não như chụp cộng hưởng từ (MRI). Dấu hiệu này do quá trình phát triển cấu trúc gần phía sau não bất thường, bao gồm thùy nhộng tiểu não và thân não.
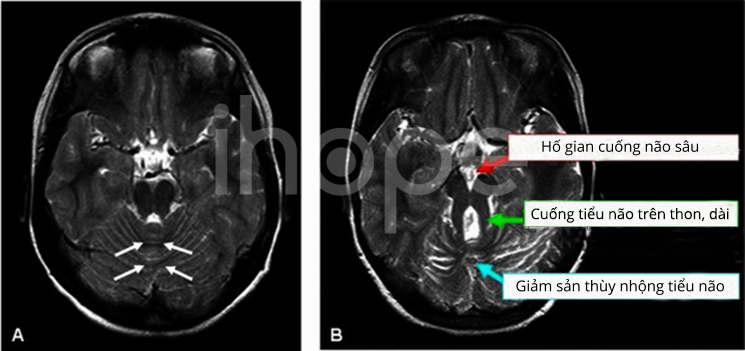
Hình A: Ảnh chụp MRI bộ não bình thường. Hình B: Ảnh chụp MRI não của người mắc hội chứng Joubert, các mũi tên chỉ ra ba thành phần của dấu hiệu răng hàm.
Nguồn: U.S National Library of Medicine
Một số xét nghiệm chẩn đoán bổ sung:
- Xét nghiệm đánh giá chức năng thận: nồng độ nitơ trong máu, nồng độ creatinin huyết thanh, công thức máu toàn phần (CBC) và phân tích nước tiểu
- Xét nghiệm đánh giá chức năng gan: nồng độ transaminase, albumin, bilirubin và thời gian prothrombin trong huyết thanh
- Chụp X-quang kiểm tra chứng loạn sản xương
- Xét nghiệm di truyền tìm đột biến gen liên quan đến bệnh
Điều trị
Hiện nay chưa có cách điều trị hội chứng Joubert. Các liệu pháp hỗ trợ dựa vào triệu chứng cụ thể mỗi người bệnh nhằm cải thiện chất lượng cuộc sống của họ:
- Vật lý trị liệu, liệu pháp ngôn ngữ hỗ trợ trẻ chậm phát triển
- Trẻ khó ăn có thể đặt ống thông dạ dày
- Trẻ có kiểu hô hấp bất thường cần theo dõi và hỗ trợ điều trị bằng thuốc kích thích hoặc bổ sung oxy hoặc mở khí quản
- Các tật khúc xạ thị giác được điều trị bằng phương pháp hiệu chỉnh thấu kính
- Người bệnh nên khám định kỳ hàng năm nhằm phát hiện và theo dõi các bất thường về gan, thận và võng mạc. Trường hợp bệnh nhân bị suy thận, suy gan, xơ gan được điều trị bằng phương pháp lọc máu hoặc phẫu thuật cấy ghép
- Bệnh ptosis hoặc mắt lác có thể dùng phương pháp phẫu thuật
Dạng di truyền
Hội chứng Joubert di truyền theo kiểu lặn trên nhiễm sắc thể thường. Do đó, bệnh chỉ biểu hiện khi có cả hai bản sao của gen đột biến trong mỗi tế bào. Bệnh nhân mắc bệnh lặn trên nhiễm sắc thể thường sẽ có bố và mẹ mang một bản sao của gen đột biến, nhưng bố mẹ ít khi biểu hiện triệu chứng bệnh.

Nguồn: U.S. National Library of Medicine
Một số trường hợp hội chứng Joubert do đột biến gen OFD1 di truyền theo kiểu lặn liên kết với nhiễm sắc thể giới tính X. Nam giới chỉ có một nhiễm sắc thể X, do đó một bản sao của gen OFD1 đột biến trong mỗi tế bào đủ gây ra suy thận và các triệu chứng nghiêm trọng khác của bệnh. Người cha bị bệnh không truyền tính trạng này cho con trai. Ở phụ nữ có hai nhiễm sắc thể X, đột biến tại một bản sao của gen OFD1 thường chỉ gây ra các triệu chứng nhẹ.
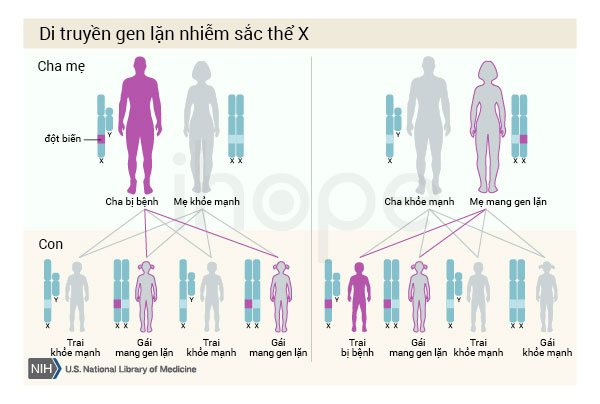
Nguồn: U.S. National Library of Medicine
Phòng ngừa
Hội chứng Joubert có cơ chế di truyền phức tạp, cha mẹ mang đột biến dị hợp nên gần như không có biểu hiện bệnh, do đó rất khó phát hiện cho đến khi sinh con. Để chủ động phòng ngừa, cha mẹ nên làm xét nghiệm sàng lọc gen lặn để chủ động cho tương lai của con. Các cặp vợ chồng trước khi mang thai cần tư vấn và xét nghiệm di truyền đảm bảo sinh con khỏe mạnh.
Các tên gọi khác
- CORS 1
- CPD4
- Cerebellar vermis agenesis
- Cerebello Oculo Renal syndrome 1
- Cerebellar Parenchymal disorder 4
- JBTS1
References
- Genetic Testing Information. Joubert syndrome. Retrieved September 11, 2022 from https://www.ninds.nih.gov/health-information/disorders/joubert-syndrome
- Genetic and Rare Diseases Information Center. Joubert syndrome. Retrieved September 11, 2022 from https://rarediseases.info.nih.gov/diseases/6802/joubert-syndrome
- Catalog of Genes and Diseases from OMIM. JOUBERT SYNDROME 1. Retrieved September 11, 2022 from https://omim.org/entry/213300
- U.S National Library of Medicine. Joubert syndrome. Retrieved September 11, 2022 from https://medlineplus.gov/genetics/condition/joubert-syndrome/
- Cleveland Clinic. Joubert Syndrome. Retrieved September 11, 2022 from https://my.clevelandclinic.org/health/articles/6040-joubert-syndrome
- National Organization for Rare Disorders. Joubert Syndrome. Retrieved September 11, 2022 from https://rarediseases.org/rare-diseases/joubert-syndrome/
- National Institute of Neurological Disorders and Stroke. Joubert Syndrome. Retrieved September 11, 2022 from https://www.ninds.nih.gov/health-information/disorders/joubert-syndrome
- Joubert Syndrome & Related Disorders Foundation. Joubert Syndrome. Retrieved September 11, 2022 from https://jsrdf.org/
- Orphanet. Joubert Syndrome. Retrieved September 11, 2022 from https://www.orpha.net/consor/cgi-bin/OC_Exp.php?Lng=GB&Expert=475
- Parisi, Melissa A. The molecular genetics of Joubert syndrome and related ciliopathies: The challenges of genetic and phenotypic heterogeneity. Published on July 4, 2019, https://doi.org/10.3233/TRD-190041
- Great Ormond Street Hospital. Joubert's syndrome. Retrieved September 11, 2022 from https://www.gosh.nhs.uk/conditions-and-treatments/conditions-we-treat/jouberts-syndrome/