Hội chứng não to-đa hồi não nhỏ-thừa ngón-não úng thuỷ (Megalencephaly-Polymicrogyria-Polydactyly-Hydrocephalus syndrome – MPPH) là bệnh lí ảnh hưởng đến quá trình phát triển não bộ, do đó gây ra nhiều triệu chứng liên quan đến thần kinh.
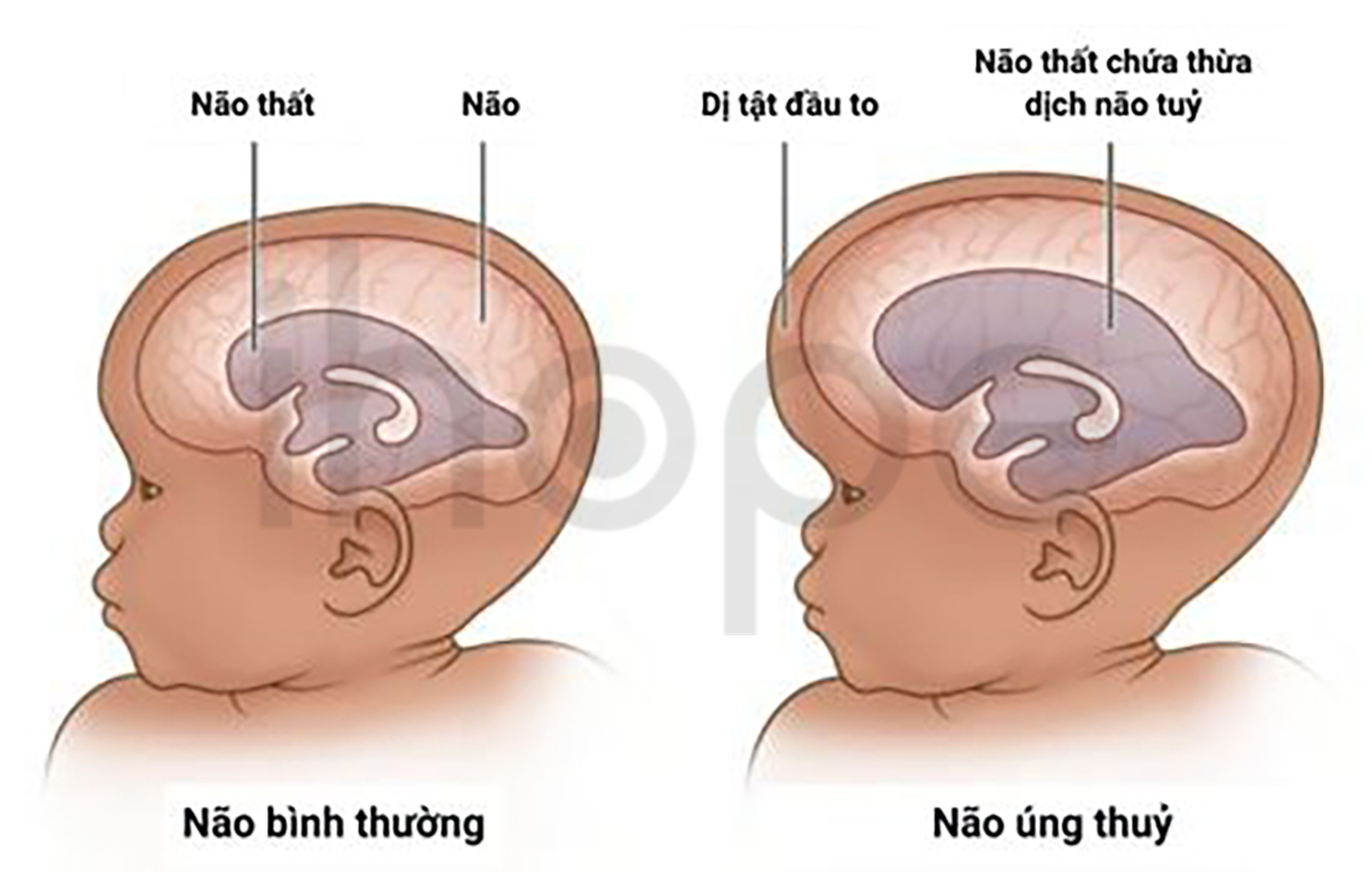
Ảnh: Não úng thuỷ
Nguồn: Zimbabwe Association of Neurological Surgeons
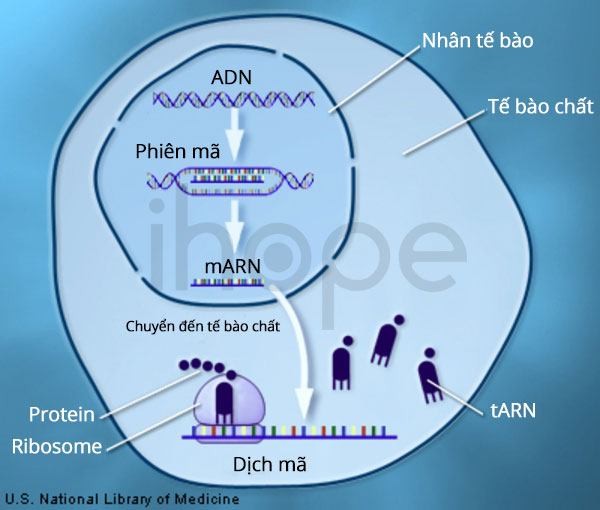
Ảnh: Quá trình sản xuất protein
Nguồn: U.S. National Library of Medicine
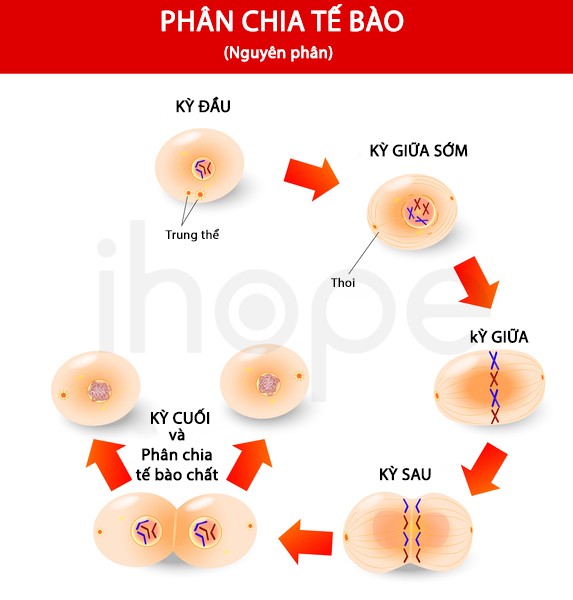
Ảnh: Quá trình phân chia tế bào (Nguyên phân)
Nguồn: Designua/Shutterstock.com
Biểu hiện lâm sàng
Trẻ mắc hội chứng não to-đa hồi não nhỏ-thừa ngón-não úng thuỷ thường có kích thước não và chu vi đầu lớn hơn bình thường do não phát triển nhanh trong hai năm đầu đời. Ngoài ra, vùng xung quanh rãnh Sylvian (đường phân cắt thuỳ trán, thuỳ đỉnh với thuỳ thái dương) xuất hiện nhiều nếp gấp có kích thước nhỏ và hình dạng bất thường được gọi là hồi não. Hơn nữa, triệu chứng não úng thuỷ đã được ghi nhận trong một số trường hợp.
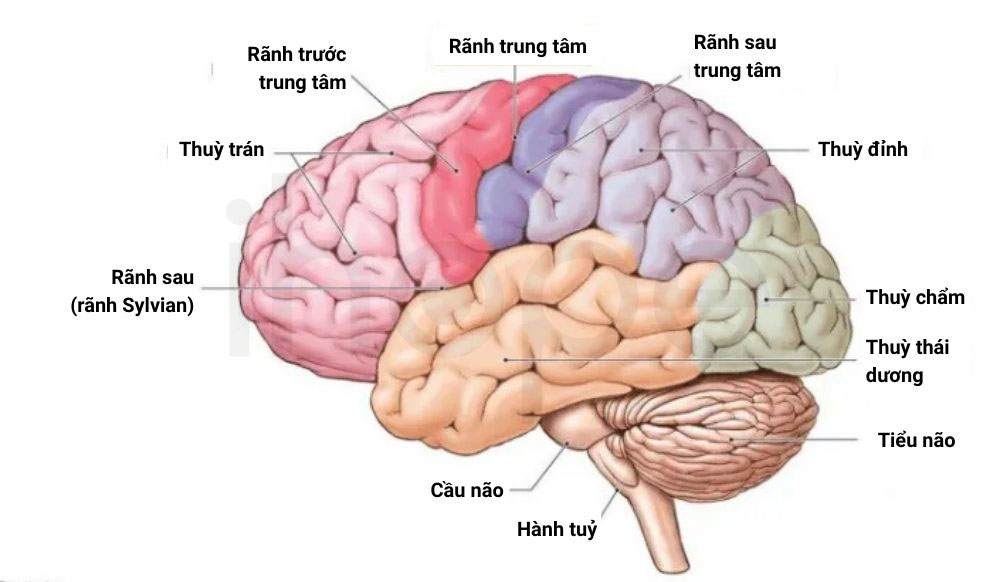
Nguồn: The Human Memory
Các biểu hiện khác của hội chứng não to-đa hồi não nhỏ-thừa ngón-não úng thuỷ bao gồm:
- Chậm phát triển và suy giảm trí tuệ mức độ nhẹ đến nặng
- Động kinh tái phát xuất hiện từ giai đoạn trẻ nhỏ
- Khó phối hợp vận động miệng và lưỡi dẫn đến chảy dãi, khó nuốt
- Chậm phát triển khả năng sử dụng ngôn ngữ
- Thừa ngón tay hoặc ngón chân
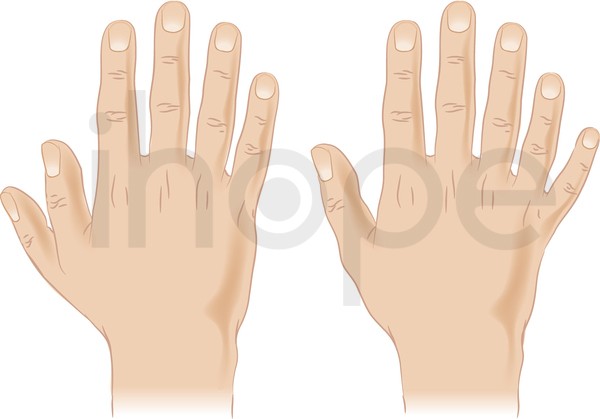
Dị tật thừa ngón tay (polydactyly) xuất hiện như một phần của tập hợp các bất thường. Polydactyly có thể tồn tại độc lập, trong trường hợp này nó được di truyền kiểu trội trên nhiễm sắc thể thường.
Nguồn: Darryl Leja, NHGRI
Độ phổ biến
Hiện nay, người ta đã ghi nhận khoảng 60 trường hợp bệnh nhân mắc hội chứng não to-đa hồi não nhỏ-thừa ngón-não úng thuỷ.
Nguyên nhân
Đột biến gen AKT3, CCND2 hoặc PIK3R2 có thể gây ra hội chứng não to-đa hồi não nhỏ-thừa ngón-não úng thuỷ. Những gen này cung cấp hướng dẫn tạo ra protein trong đường tín hiệu PI3K-AKT-mTOR—tín hiệu điều phối nhiều hoạt động thiết yếu của tế bào, bao gồm tổng hợp protein mới , tăng trưởng và phân chia tế bào cũng như duy trì khả năng sống của tế bào. Vì vậy, những protein này rất quan trọng đối với quá trình phát triển bình thường của nhiều cơ quan, đặc biệt là não bộ.
Đột biến một trong các gen này có thể khiến protein hoạt động quá mức hoặc protein không được phân huỷ đúng thời điểm. Do đó, tín hiệu PI3K-AKT-mTOR hoạt động mạnh hơn dẫn đến các tế bào tăng sinh và phát triển quá nhanh. Số lượng tế bào tăng nhanh bất thường ảnh hưởng đến cấu trúc và chức năng của não bộ từ lúc não đang hình thành trong giai đoạn thai nhi.
Người ta chưa hiểu rõ vì sao đột biến dẫn đến triệu chứng thừa ngón nhưng dự đoán nó liên quan đến hiện tượng tăng sinh bất thường của tế bào trong quá trình phát triển bàn tay và bàn chân.
Chẩn đoán
Hội chứng não to-đa hồi não nhỏ-thừa ngón-não úng thuỷ được chẩn đoán dựa trên các phương pháp sau:
- Đánh giá lâm sàng: quan sát những triệu chứng liên quan đến phát triển thần kinh, vận động và các đặc điểm bất thường quanh vùng đầu
- Chụp cộng hưởng từ: phát hiện não úng thủy hoặc các dị tật não khác
- Đo điện não: xác định những bất thường trong hoạt động điện não và triệu chứng co giật, động kinh
- Xét nghiệm di truyền: xác định đột biến gen AKT3, CCND2 hoặc PIK3R2 nhằm xác nhận chẩn đoán

Nguồn: verywellhealth.com
Điều trị
Hiện nay, chưa có phương pháp điều trị hoàn toàn hội chứng não to-đa hồi não nhỏ-thừa ngón-não úng thuỷ. Các liệu pháp chủ yếu nhằm giảm nhẹ triệu chứng và cải thiện chất lượng đời sống người bệnh. Trẻ bệnh cần được theo dõi thường xuyên nhằm giám sát các triệu chứng và quá trình tiến triển của bệnh. Trong một số trường hợp, bác sĩ có thể kê đơn thuốc chống động kinh nhằm điều trị co giật. Ngoài ra, trị liệu ngôn ngữ, vật lí trị liệu có thể cải thiện các hoạt động sinh hoạt hàng ngày của bệnh nhân.
Dạng di truyền
Phần lớn các trường hợp mắc hội chứng não to-đa hồi não nhỏ-thừa ngón-não úng thuỷ do đột biến mới (de novo) xuất hiện trong quá trình hình thành tế bào sinh sản (trứng, tinh trùng) hoặc quá trình phát triển phôi sớm. Các trường hợp di truyền bệnh từ cha mẹ hiếm gặp hơn.
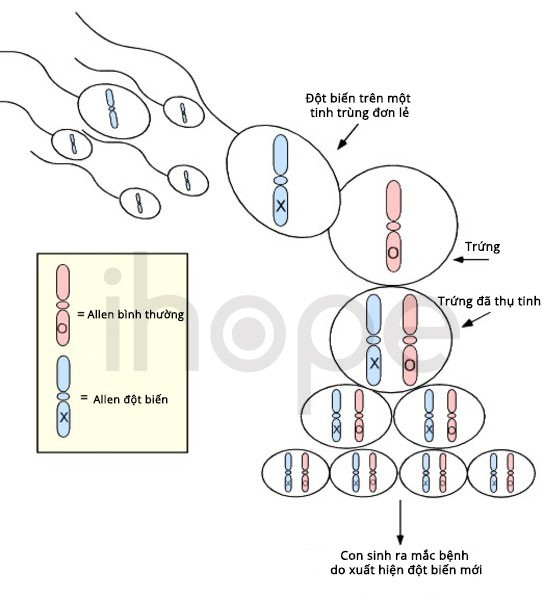
Nguồn: U.S. National Library of Medicine
Phòng ngừa
Hội chứng não to-đa hồi não nhỏ-thừa ngón-não úng thuỷ bắt nguồn từ đột biến ngẫu nhiên xảy ra trong quá trình tạo phôi, vì vậy tất cả thai phụ đều có nguy cơ mang thai bị bệnh. Do đó, các cặp vợ chồng trước khi mang thai cần tư vấn và xét nghiệm di truyền nhằm đảm bảo sinh con khỏe mạnh. Đồng thời, thai phụ cần theo dõi chặt chẽ thai kì bằng siêu âm và các xét nghiệm sàng lọc như NIPT cũng như thăm khám định kì với bác sĩ sản khoa. Người thuộc nhóm nguy cơ mắc bệnh cần khám sức khỏe và tầm soát bệnh định kì.
Các tên gọi khác
- MEG-PMG-POLY-HYD
- Megalencephaly-postaxial polydactyly-polymicrogyria-hydrocephalus syndrome
- MPPH
- MPPH syndrome
References
- National Library of Medicine. Megalencephaly-polymicrogyria-polydactyly-hydrocephalus syndrome 1. Retrieved September 2, 2025 from https://www.ncbi.nlm.nih.gov/gtr/conditions/C4012727/
- National Organisation for Rare Disorders. Megalencephaly-polymicrogyria-postaxial polydactyly-hydrocephalus syndrome. Retrieved September 2, 2025 from https://rarediseases.org/mondo-disease/megalencephaly-polymicrogyria-postaxial-polydactyly-hydrocephalus-syndrome/
- OMIM. MEGALENCEPHALY-POLYMICROGYRIA-POLYDACTYLY-HYDROCEPHALUS SYNDROME 1; MPPH1. Retrieved September 2, 2025, from https://omim.org/entry/603387
- U.S National Library of Medicine. Megalencephaly-polymicrogyria-polydactyly-hydrocephalus syndrome. Retrieved September 2, 2025 from https://medlineplus.gov/genetics/condition/megalencephaly-polymicrogyria-polydactyly-hydrocephalus-syndrome/
- Orphanet. Megalencephaly-polymicrogyria-postaxial polydactyly-hydrocephalus syndrome. Retrieved September 2, 2025 from https://www.orpha.net/en/disease/detail/83473
- National Library of Medicine. MPPH Syndrome. Retrieved September 2, 2025 from https://www.ncbi.nlm.nih.gov/books/NBK396098/