Hội chứng miệng-mặt-ngón (oral-facial-digital syndrome) là hội chứng di truyền hiếm gặp. Người ta ghi nhận 14 dạng hội chứng miệng-mặt-ngón khác nhau, trong đó hội chứng dạng I là phổ biến nhất. Một số biểu hiện đặc trưng của hội chứng bao gồm khoang miệng kém phát triển, khuôn mặt dị biệt và dị tật ngón tay.
Biểu hiện lâm sàng
Người ta phân loại hội chứng miệng-mặt-ngón dựa trên các triệu chứng nhưng phần lớn bệnh nhân đều biểu hiện những dấu hiệu sau:
Dị tật khoang miệng
Khoang miệng của người bệnh không phát triển hoàn thiện, dẫn đến các biểu hiện như:
- Hở môi , hở hàm ếch
- Lưỡi chẻ đôi, chia thuỳ hoặc có khối u
- Răng thừa hoặc thiếu
- Hàm nhỏ
- Lệch khớp cắn
- Môi và nướu dính nhau
- Nhiễm trùng đường hô hấp
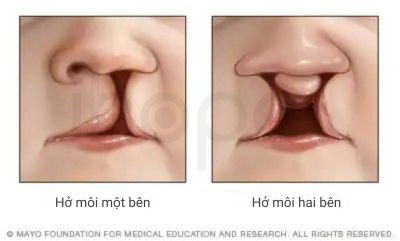
Ảnh: Hở môi
Nguồn: Mayo Clinic

Ảnh: Hở hàm ếch
Nguồn: Health Centers for Disease Control and Prevention
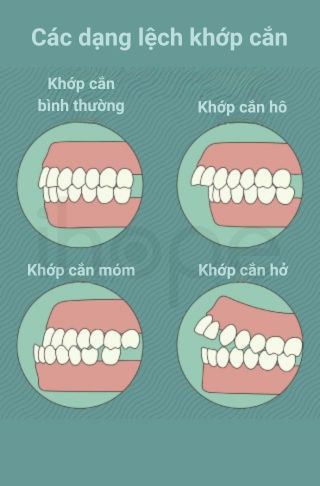
Ảnh: Các dạng lệch khớp cắn
Nguồn: Chats Dental
Khuôn mặt dị biệt
Bệnh nhân có những đặc điểm dị biệt trên khuôn mặt như:
- Hai mắt xa nhau
- Mắt lác
- Kích thước lỗ mũi không đều
- Rụng tóc thành từng mảng
- Mũi to và phẳng
- Tai góc cạnh
Dị tật tay-chân
Hội chứng ảnh hưởng đến quá trình phát triển xương của trẻ từ trong bụng mẹ, dẫn đến các dị tật như:
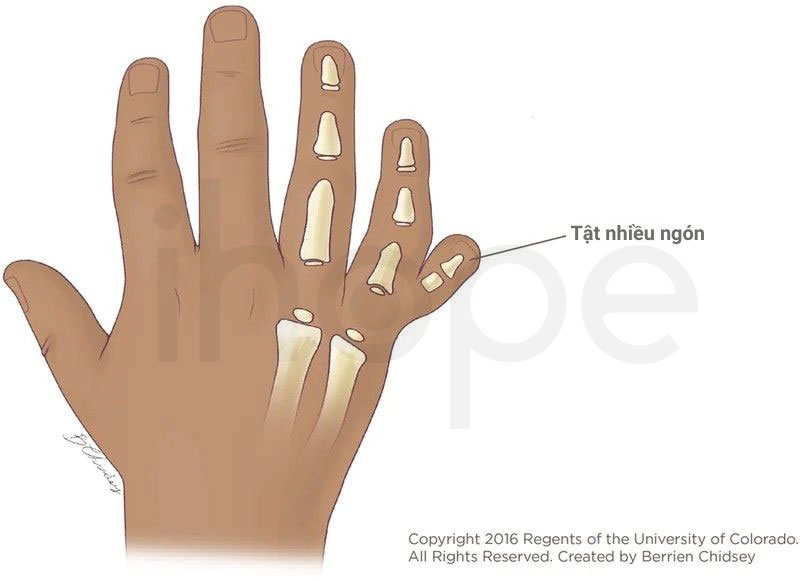
Ảnh: Tật nhiều ngón
Nguồn: Children's Hospital Colorado
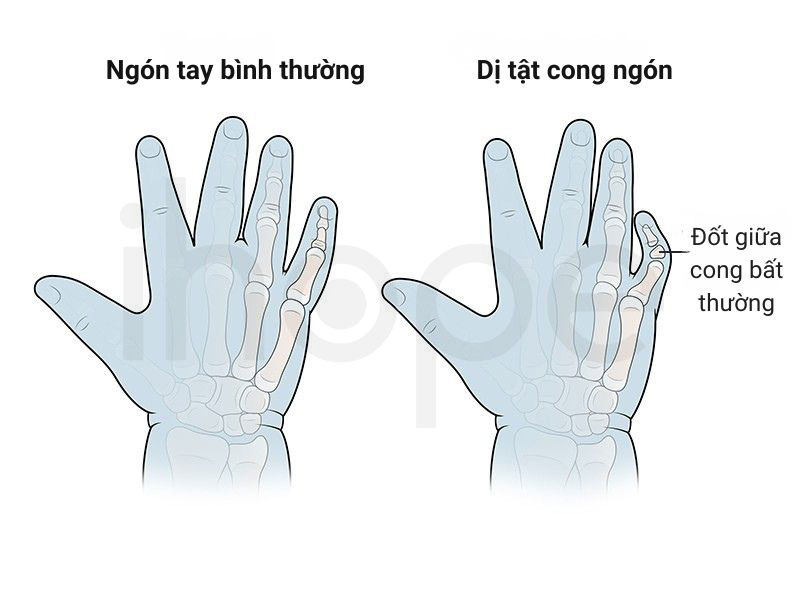
Ảnh: Dị tật cong ngón tay
Nguồn: Boston Children's Hospital
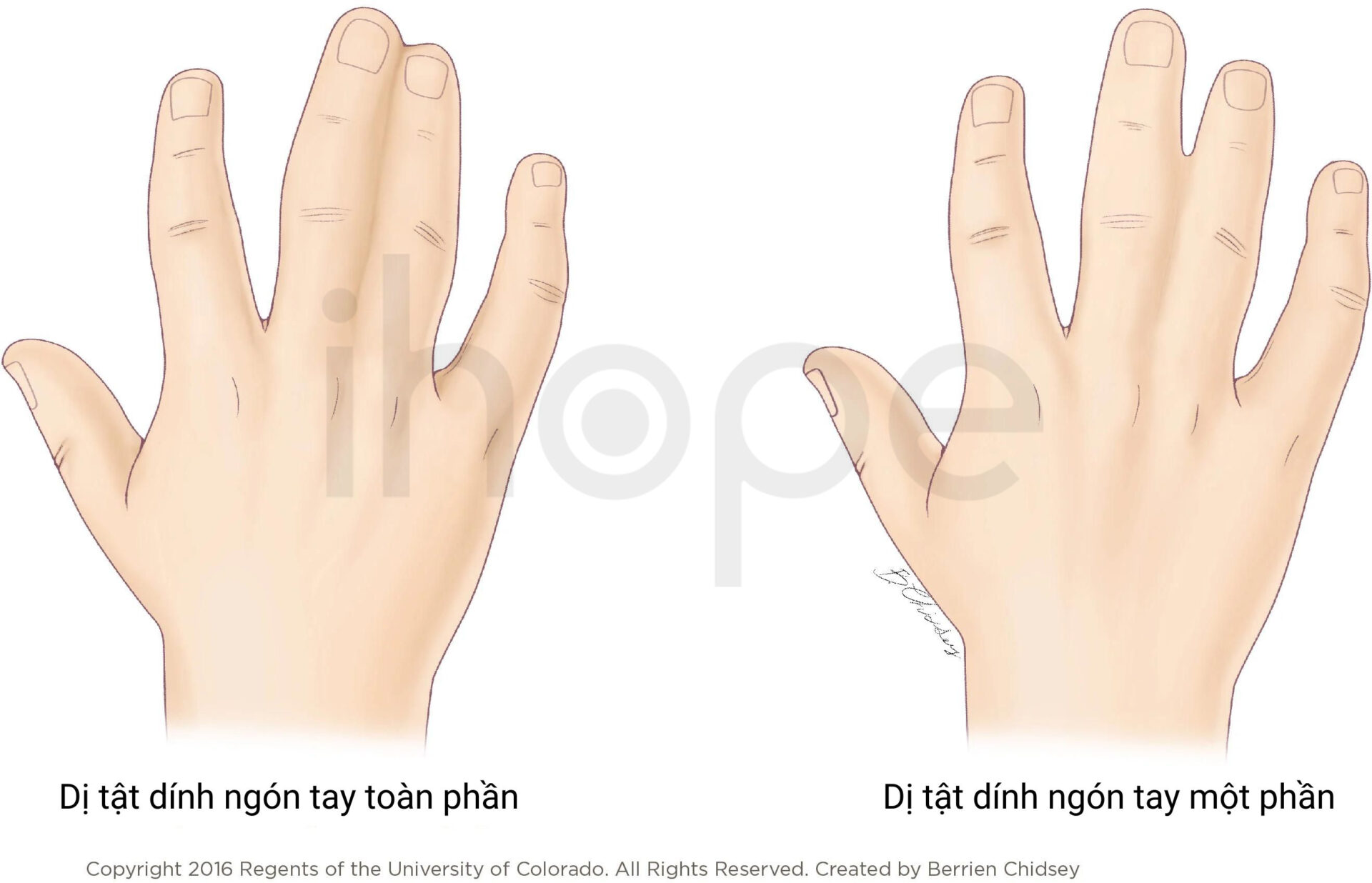
Ảnh: Dị tật dính ngón tay
Nguồn: Children's Hospital Colorado

Ảnh: Bàn chân khoèo
Nguồn: U.S. National Library of Medicine
Bệnh lí về não-thần kinh
Hội chứng gây ra một số bất thường về não bộ và hành vi như:
- Thiểu năng trí tuệ
- Co giật
- Chậm nói
- Giảm khả năng vận động
- Chậm lớn
Độ phổ biến
Ước tính tỉ lệ mắc hội chứng miệng-mặt-ngón tay dạng I khoảng 1/50.000–250.000 trẻ. Tỉ lệ mắc hội chứng dạng II khoảng 1/300.000 trẻ. Các dạng hội chứng miệng-mặt-ngón còn lại rất hiếm gặp nên tỉ lệ mắc bệnh chưa được xác định cụ thể.
Nguyên nhân
Hội chứng miệng-mặt-ngón bắt nguồn từ đột biến gen OFD1 trên nhiễm sắc thể X. Gen OFD1 cung cấp hướng dẫn tạo ra protein OFD1 trong lông mao—cấu trúc cực nhỏ nhô ra khỏi bề mặt tế bào. Lông mao tham gia vào nhiều đường tín hiệu giữa các tế bào, do đó nó giữ vai trò quan trọng đối với quá trình phát triển và chức năng của nhiều mô, đặc biệt là thận, gan, võng mạc. Ngoài ra, lông mao cũng tham gia vào quá trình phát triển xương.
Đột biến gen OFD1 làm thay đổi cấu trúc protein OFD1 nên protein hoạt động không như bình thường. Do đó, cấu trúc lông mao không được hoàn thiện, dẫn đến xương, mô ngoại bì cùng các mô và cơ quan khác không thể phát triển bình thường.
Mỗi dạng hội chứng miệng-mặt-ngón có liên quan đến một gen hoặc nhiều gen khác như NEK1, TMEM231, TCTN3, WDPCP, DDX59, TMEM216, MEM107, KIAAO753,... Đa số các gen này đều tham gia vào quá trình hình thành và hoạt động của lông mao.

Nguồn: LibreTexts Health
Chẩn đoán
Hội chứng miệng-mặt-ngón được chẩn đoán dựa trên biểu hiện lâm sàng, bệnh sử gia đình kết hợp với kết quả xét nghiệm sinh hóa, hình ảnh và di truyền.
Xét nghiệm sinh hoá
Các xét nghiệm sinh hóa cung cấp thông tin về chức năng gan, thận, nội tiết để bác sĩ có thể theo dõi các biến chứng liên quan đến tim hoặc hệ thần kinh. Bệnh nhân được chỉ định những xét nghiệm khác nhau nhằm phân biệt hội chứng miệng-mặt-ngón với các bệnh lí di truyền khác và xây dựng phác đồ điều trị hiệu quả.
Chẩn đoán hình ảnh
Các phương pháp chẩn đoán hình ảnh như MRI, chụp cắt lớp và siêu âm giúp bác sĩ phát hiện những đặc điểm bất thường trong quá trình phát triển của trẻ.
Xét nghiệm di truyền
Bác sĩ thực hiện các xét nghiệm di truyền nhằm phát hiện đột biến gen gây ra hội chứng, qua đó kết quả chẩn đoán được xác nhận.
Điều trị
Hiện nay, chưa có phương pháp điều trị hoàn toàn hội chứng miệng-mặt-ngón. Các liệu pháp chủ yếu nhằm giảm nhẹ triệu chứng và cải thiện chất lượng đời sống người bệnh.
Một số liệu pháp hỗ trợ bao gồm:
- Phẫu thuật điều trị dị tật
- Sử dụng thuốc chống co giật
- Trị liệu ngôn ngữ, hành vi
- Vật lí trị liệu
Dạng di truyền
Hội chứng miệng-mặt-ngón dạng I và VII di truyền theo kiểu trội trên nhiễm sắc thể giới tính X. Nam giới chỉ có một nhiễm sắc thể X, do đó một bản sao của gen đột biến trong mỗi tế bào đủ gây ra triệu chứng bệnh nghiêm trọng. Người cha bị bệnh không thể truyền gen đột biến này cho con trai. Phụ nữ có hai nhiễm sắc thể X, đột biến tại một bản sao của gen thường chỉ gây ra các triệu chứng nhẹ.
Hội chứng dạng II, III, IV, V, VI, IX, IV di truyền theo kiểu lặn trên nhiễm sắc thể thường. Do đó, bệnh chỉ biểu hiện khi có cả hai bản sao của gen đột biến trong mỗi tế bào. Bệnh nhân mắc bệnh lặn trên nhiễm sắc thể thường sẽ có bố và mẹ mang một bản sao của gen đột biến, nhưng họ ít khi biểu hiện triệu chứng bệnh.

Nguồn: U.S. National Library of Medicine
Phòng ngừa
Hội chứng miệng-mặt-ngón có cơ chế di truyền phức tạp nên khó phát hiện ở các cặp cha mẹ mang gen bệnh cho đến khi sinh con. Để chủ động phòng ngừa, cha mẹ nên làm xét nghiệm sàng lọc gen lặn cho tương lai của con. Các cặp vợ chồng trước khi mang thai cần tư vấn và xét nghiệm di truyền đảm bảo sinh con khỏe mạnh. Người thuộc nhóm nguy cơ mắc bệnh cần khám sức khỏe và tầm soát bệnh định kì.
Các tên gọi khác
- Dysplasia linguofacialis
- OFDS
- Oro-facio-digital syndrome
- Orodigitofacial dysostosis
- Orodigitofacial syndrome
- Orofaciodigital syndrome
References
- MedlinePlus. Oral-facial-digital syndrome. Retrieved July 25, 2025 from https://medlineplus.gov/genetics/condition/oral-facial-digital-syndrome/
- National Organization for Rare Disorder. Oral-Facial-Digital Syndrome. Retrieved July 25, 2025 from https://rarediseases.org/rare-diseases/oral-facial-digital-syndrome/
- Orphanet. Orofaciodigital syndrome type 1. Retrieved July 25, 2025 from https://www.orpha.net/en/disease/detail/2750
- National Human Genome Research Institute. Oral-Facial-Digital Syndromes (OFDS) Research Study. Retrieved July 25, 2025 from https://www.genome.gov/27529974/ofd-general-information
- Radiopaedia. Oral-facial-digital syndromes. Retrieved July 25, 2025 from https://radiopaedia.org/articles/oral-facial-digital-syndromes-1





