Loạn dưỡng mỡ (lipodystrophy) là bệnh hiếm gặp với biểu hiện thiếu mô mỡ toàn thân. Mô mỡ hiện diện tại nhiều nơi trên cơ thể, bao gồm bên dưới da và xung quanh các cơ quan nội tạng. Chúng dự trữ chất béo để cung cấp năng lượng cho cơ thể, đồng thời đóng vai trò như lớp đệm. Rối loạn phân bố mỡ toàn thân bẩm sinh thuộc nhóm nhóm các rối loạn dưỡng mỡ. Người bệnh bị mất mô mỡ, nên chất béo được lưu trữ tại những nơi khác trong cơ thể ví dụ trong gan và cơ, từ đó phát sinh các vấn đề sức khỏe nghiêm trọng.
Bệnh được chia thành hai dạng dựa vào nguyên nhân di truyền:
- Loạn dưỡng mỡ toàn thân bẩm sinh (congenital generalized lipodystrophy)
- Loạn dưỡng mỡ một phần gia đình (familial partial lipodystrophy)
Biểu hiện lâm sàng
Người bệnh bị mất mô mỡ ở vùng cánh tay, chân và hông, khiến những bộ phận này trông rắn chắc và cơ bắp. Chất béo tích tụ quanh mặt, cổ và bên trong bụng tạo ra dáng người "cushingoid" vì mỡ thừa tại những khu vực này mang lại vẻ ngoài giống với các đặc điểm của hội chứng Cushing. Quá trình phân bố chất béo bất thường có thể xuất hiện bất cứ lúc nào, ngay sau sinh cho đến tuổi trưởng thành.
Nhiều bệnh nhân mắc phải tình trạng kháng insulin. Các mô trong cơ thể không thể đáp ứng đầy đủ với insulin - hormone điều hòa lượng đường trong máu. Tình trạng kháng insulin kéo dài gây ra bệnh tiểu đường.
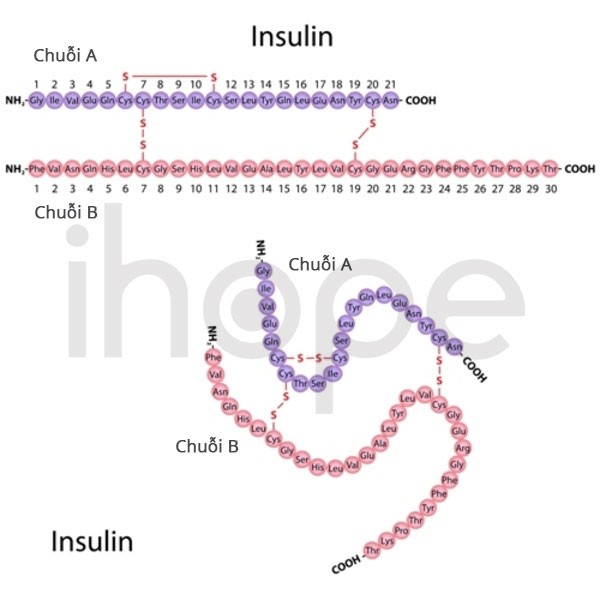
Nguồn: U.S. National Library of Medicine
Hầu hết, người bệnh có chất béo trung tính triglyceride trong máu cao dẫn đến viêm tuyến tụy. Ngoài ra, chứng loạn dưỡng mỡ toàn thân bẩm sinh gây tích tụ chất béo trong gan (gan nhiễm mỡ), dẫn đến gan to và suy gan. Da của họ dày, sẫm màu và mịn như nhung tại các nếp gấp trên cơ thể, tình trạng này được gọi là chứng acanthosis nigrican. Sau tuổi dậy thì, một số phụ nữ bị bệnh phát triển u nang buồng trứng, nhiều lông trên cơ thể và vô sinh liên quan đến thay đổi nội tiết tố.
Người ta đã tìm hiểu được ít nhất sáu loại phụ của loạn dưỡng mỡ một phần gia đình dựa trên nguyên nhân di truyền. Dạng bệnh phổ biến nhất là loại 2, còn gọi là bệnh Dunnigan. Ngoài các dấu hiệu và triệu chứng được mô tả ở trên, một số người bệnh phát triển chứng yếu cơ, bệnh cơ tim, bệnh động mạch vành và các vấn đề với hệ thống dẫn truyền và điều phối nhịp tim.
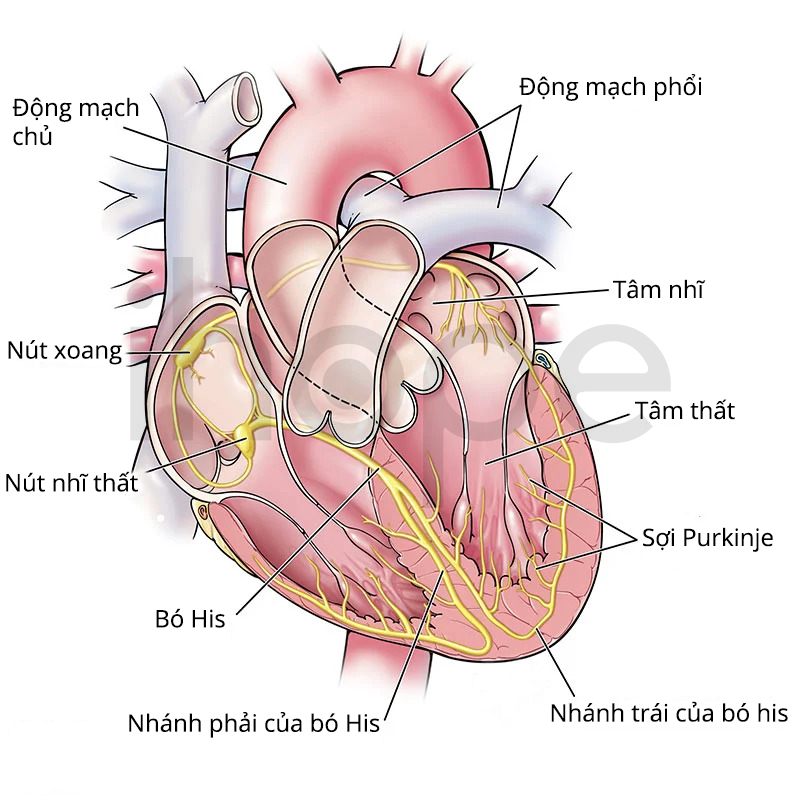
Nguồn: Cleveland Clinic
Độ phổ biến
Loạn dưỡng mỡ một phần gia đình là bệnh hiến gặp. Tỷ lệ mắc chứng loạn dưỡng mỡ một phần gia đình ước tính khoảng 1/1.000.000 người. Loại 2 là dạng phổ biến nhất với hơn 500 trường hợp được báo cáo trong các tài liệu y khoa. Phụ nữ có nguy cơ mắc bệnh cao hơn nam giới, nguyên nhân có thể do giảm mỡ hông và tay chân ở phụ nữ dễ nhận ra, bên cạnh đó các biến chứng như tiểu đường và tăng triglycerid máu xảy ra phổ biến hơn.
Nguyên nhân
Loạn dưỡng mỡ một phần trong gia đình do đột biến tại một số gen bao gồm LMNA, PPARG, PLIN1, AKT2 VÀ CIDEC. Trong đó, đột biến gen LMNA gây ra loại 2, các loại khác ít gặp.
Các gen khác liên quan đến chứng loạn dưỡng mỡ một phần gia đình cung cấp hướng dẫn tạo ra protein với nhiều chức năng khác nhau bao gồm lưu trữ chất béo. Cụ thể hơn, các protein này giữ vai trò quan trọng trong quá trình phát triển và hoạt động của tế bào mỡ - tế bào dự trữ chất béo trong mô mỡ.
Đột biến tại bất kỳ gen nào liên quan đến chứng loạn dưỡng mỡ một phần gia đình làm giảm hoặc mất hoàn toàn chức năng của các protein tương ứng, từ đó gây suy giảm quá trình phát triển, cấu trúc hoặc chức năng của tế bào mỡ, do đó cơ thể không thể lưu trữ và sử dụng chất béo như bình thường. Những bất thường này của mô mỡ làm thay đổi quá trình sản xuất hormone và ảnh hưởng đến nhiều cơ quan trong cơ thể. Tuy nhiên, người ta chưa hiểu rõ vì sao gen đột biến khiến chất béo bị mất đi tại một số bộ phận và tích trữ bất thường trong những bộ phận khác.
Một số người bệnh không mang đột biến trong các gen đã biết. Các nhà nghiên cứu đang tìm kiếm những biến đổi di truyền khác liên quan đến bệnh.
Chẩn đoán
Người bệnh được khám sức khỏe và thăm hỏi tiền sử bệnh gia đình. Người bệnh ốm hoặc không thừa cân nhưng mắc bệnh tiểu đường sớm, tăng triglycerid máu nặng, gan nhiễm mỡ, gan lách to, acanthosis nigrican, buồng trứng đa nang có nguy cơ cao mắc loạn dưỡng mỡ toàn thân bẩm sinh.
Một số xét nghiệm chẩn đoán như:
- Xét nghiệm máu kiểm tra nồng độ glucose, lipid, men gan và axit uric.
- Kết quả xét nghiệm hình ảnh như chụp cộng hưởng từ (MRI) cho thấy mất chất béo.
- Xét nghiệm di truyền tìm đột biến gen gây loạn dưỡng mỡ toàn thân bẩm sinh.
Điều trị
Hiện nay chưa có phương pháp điều trị hoàn toàn loạn dưỡng mỡ toàn thân bẩm sinh. Các liệu pháp tập trung vào triệu chứng cụ thể và cải thiện chất lượng cuộc sống của người bệnh.
- Thực hiện chế độ ăn nhiều carbohydrate, ít chất béo
- Tập thể dục thường xuyên, duy trì cân nặng hợp lý
Bệnh nhân cần được khám tim định kỳ nhằm phát hiện biến chứng kịp thời. Các bất thường về tim như block tim hoặc rung tâm nhĩ có thể yêu cầu sử dụng máy tạo nhịp tim. Ghép tim đối với trường hợp suy tim nghiêm trọng.
Dạng di truyền
Hầu hết, các trường hợp loạn dưỡng mỡ một phần gia đình di truyền theo kiểu trội trên nhiễm sắc thể thường. Do đó, một bản sao của gen đột biến trong mỗi tế bào đủ gây ra bệnh. Người bệnh có thể thừa hưởng đột biến từ cha hoặc mẹ bị bệnh.

Nguồn: U.S. National Library of Medicine
Một số trường hợp do đột biến gen mới và xảy ra ở những người không có tiền sử mắc bệnh trong gia đình họ.
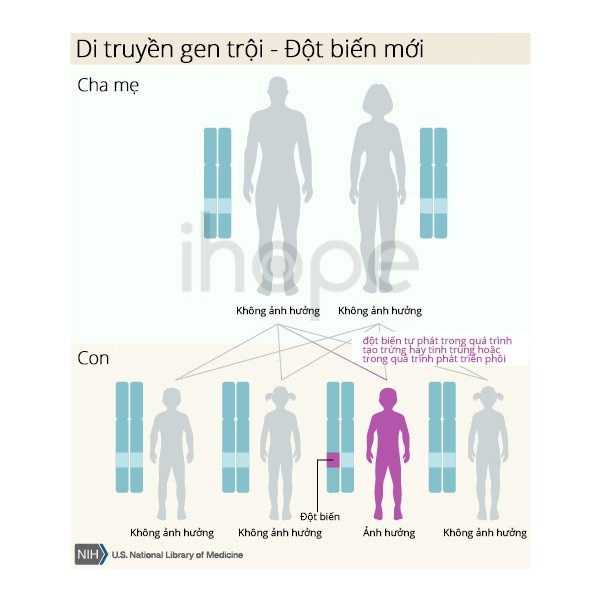
Nguồn: U.S. National Library of Medicine
Các trường hợp hiếm gặp của bệnh loạn dưỡng mỡ một phần gia đình di truyền theo kiểu lặn trên nhiễm sắc thể thường. Do đó, bệnh chỉ biểu hiện khi có cả hai bản sao của gen đột biến trong mỗi tế bào. Bệnh nhân mắc bệnh lặn trên nhiễm sắc thể thường sẽ có bố và mẹ mang một bản sao của gen đột biến, nhưng bố mẹ ít khi biểu hiện triệu chứng bệnh.

Nguồn: U.S. National Library of Medicine
Phòng ngừa
Loạn dưỡng mỡ một phần gia đình di truyền trội trên nhiễm sắc thể thường, khi sinh con sẽ có 50% khả năng di truyền bệnh. Do đó, để bảo đảm 100% khả năng con không bị bệnh, cha mẹ có thể chọn phương pháp thụ tinh nhân tạo IVF và sàng lọc phôi PGS/PGD.
Trường hợp bệnh di truyền lặn trên nhiễm sắc thể thường, cha mẹ mang đột biến dị hợp nên gần như không có biểu hiện bệnh, do đó rất khó phát hiện cho đến khi sinh con. Để chủ động phòng ngừa, cha mẹ nên làm xét nghiệm sàng lọc gen lặn để chủ động cho tương lai của con.
Các thành viên trong gia đình nên khám sức khỏe và tầm soát bệnh định kỳ nếu có thành viên mắc bệnh.
Các tên gọi khác
- Dunnigan-Kobberling syndrome
- FPL
- Kobberling-Dunnigan syndrome
- Lipodystrophy, familial partial
References
- Genetic Testing Information. Familial partial lipodystrophy. Retrieved September 15, 2022 from https://www.ncbi.nlm.nih.gov/gtr/conditions/C0271694/
- Genetic and Rare Diseases Information Center. Familial partial lipodystrophy. Retrieved September 15, 2022 from https://rarediseases.info.nih.gov/diseases/11962/familial-partial-lipodystrophy/
- Genetic and Rare Diseases Information Center. Familial partial lipodystrophy associated with PPARG mutations. Retrieved September 15, 2022 from https://rarediseases.info.nih.gov/diseases/12600/familial-partial-lipodystrophy-associated-with-pparg-mutations
- Catalog of Genes and Diseases from OMIM. LIPODYSTROPHY, FAMILIAL PARTIAL, TYPE 1. Retrieved September 15, 2022 from https://omim.org/entry/608600
- U.S National Library of Medicine. Familial Partial Lipodystrophy. Retrieved September 15, 2022 from https://medlineplus.gov/genetics/condition/familial-partial-lipodystrophy/
- National Organization for Rare Disorders. Familial Partial Lipodystrophy. Retrieved September 15, 2022 from https://rarediseases.org/rare-diseases/familial-partial-lipodystrophy/
- Mayo Foundation for Medical Education and Research. Familial partial lipodystrophy — one size does not fit all: Cases from the endocrine teaching clinic. Retrieved September 15, 2022 from https://www.mayoclinic.org/medical-professionals/endocrinology/news/familial-partial-lipodystrophy-one-size-does-not-fit-all-cases-from-the-endocrine-teaching-clinic/mcc-20472117
- Orphanet. Familial partial lipodystrophy. Retrieved September 15, 2022 from https://www.orpha.net/consor/cgi-bin/OC_Exp.php?lng=EN&Expert=98306
- Carolina Cecchetti, M. Rosaria D’Apice, Elena Morini, Giuseppe Novelli, Carmine Pizzi, Uberto Pagotto, Alessandra Gambineri. Case Report: An Atypical Form of Familial Partial Lipodystrophy Type 2 Due to Mutation in the Rod Domain of Lamin A/C. Published on April 19, 2021 from https://doi.org/10.3389/fendo.2021.675096
- Christos Bagias, Angeliki Xiarchou, Alexandra Bargiota and Stelios Tigas. Familial Partial Lipodystrophy (FPLD): Recent Insights. Published on May 6, 2020 from https://doi.org/10.2147/DMSO.S206053