
Hội chứng Adams-Oliver là dị tật bẩm sinh hiếm gặp với biểu hiện đặc trưng gồm bất sản biểu bì da đầu và dị tật các chi. Trẻ mắc bệnh có thể phát triển những biến chứng liên quan đến mắt, tim và thần kinh.
Biểu hiện lâm sàng
Hầu hết người mắc hội chứng Adams-Oliver gặp bất thường trong quá trình phát triển da. Họ có những vùng da bị mất trên đỉnh đầu, một số trường hợp có xương dưới da kém phát triển. Người bệnh thường có sẹo và lông không mọc ở vùng này. Những dị tật ở bàn tay và bàn chân phổ biến như tật dính ngón , tật thừa ngón hoặc thiếu ngón .
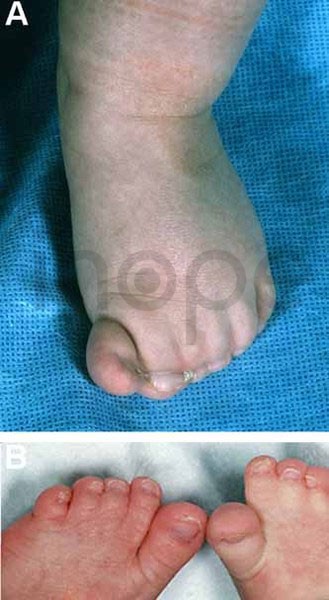
Ảnh: Tật dính ngón
Nguồn: U.S. National Library of Medicine
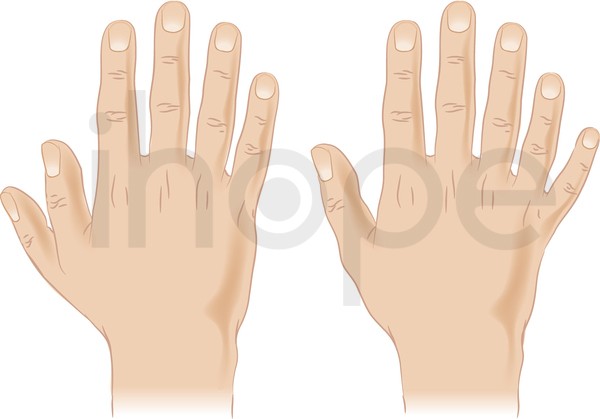
Ảnh:Dị tật thừa ngón tay
Dị tật thừa ngón tay (polydactyly) xuất hiện như một phần của tập hợp các bất thường. Polydactyly có thể tồn tại độc lập, trong trường hợp này nó được di truyền kiểu trội trên nhiễm sắc thể thường.
Nguồn: Darryl Leja, NHGRI

Ảnh: Thiếu ngón
Nguồn: U.S National Library of Medicine
Trẻ sơ sinh mắc bệnh có thể phát triển bệnh rối loạn mạch máu bẩm sinh (cutis marmorata telangiectatica congenita). Bệnh gây ra hình dạng như lưới màu đỏ hoặc tím trên da. Ngoài ra, người bệnh phát triển chứng tăng áp động mạch phổi (cao huyết áp) hoặc dị tật tim đe dọa đến tính mạng.
Một số đặc điểm khác của bệnh bao gồm:
- Vấn đề về thần kinh, thiểu năng trí tuệ
- Phát triển chậm
- Bất thường cấu trúc não
Độ phổ biến
Hội chứng Adams-Oliver hiếm gặp, do đó hiện nay tỷ lệ mắc bệnh vẫn chưa được thống kê cụ thể.
Nguyên nhân
Các đột biến gen ARHGAP31, DLL4, DOCK6, EOGT, NOTCH1 hoặc RBPJ có thể gây ra hội chứng Adams-Oliver. Những gen này đóng vai trò quan trọng trong giai đoạn phát triển phôi thai. Thay đổi xảy ra tại bất kỳ gen nào trong số này có thể làm giảm hiệu quả kiểm soát quá trình phát triển, dẫn đến dấu hiệu và triệu chứng của bệnh.
Protein tạo ra từ các gen ARHGAP31 và DOCK6 thuộc họ enzyme GTPase. Chúng hoạt động như các “công tắc”, trong đó protein DOCK6 bật và protein ARHGAP31 tắt các tín hiệu từ nhiều nguồn bên ngoài tế bào. Những protein này tham gia vào nhiều giai đoạn phát triển của phôi thai, đặc biệt tại các chi, hộp sọ và tim. Đột biến gen ARHGAP31 và DOCK6 dẫn đến các phiên bản protein ngắn bất thường và mất chức năng, từ đó protein bị giảm hoạt động. Do đó, người bệnh có các vấn đề về da, dị dạng xương và những đặc điểm khác của hội chứng Adams-Oliver.
Protein tạo ra từ các gen NOTCH1, DLL4 và RBPJ là một phần của đường tín hiệu Notch. Tín hiệu Notch kiểm soát cách một số loại tế bào phát triển trong phôi thai đang hình thành, bao gồm cả những tế bào tạo nên xương, tim, cơ, dây thần kinh và mạch máu. Các protein Notch1 và DLL4 lắp với nhau như ổ khóa và chìa, chúng kích thích một phần của đường tín hiệu Notch. Các đột biến gen NOTCH1 và DLL4 liên quan đến hội chứng Adams-Oliver làm suy giảm tín hiệu Notch1, từ đó gây ra các bất thường về mạch máu và tim với một số người bệnh. Người ta cho rằng các đặc điểm khác của bệnh có thể do mạch máu phát triển bất thường trước khi sinh.
Quá trình truyền tín hiệu thông qua Notch1 và các protein Notch khác sẽ kích thích protein RBP-J (tạo ra từ gen RBPJ) liên kết vào một số vùng ADN để kiểm soát hoạt động của các gen tham gia phát triển tế bào trong nhiều mô trên khắp cơ thể. Các đột biến gen RBPJ liên quan đến hội chứng Adams-Oliver làm thay đổi vùng liên kết với ADN của protein RBP-J, nên protein không thể liên kết với ADN để kích hoạt gen. Do đó, quá trình phát triển bình thường của da, xương và các mô khác bị gián đoạn, dẫn đến các đặc điểm của hội chứng Adams-Oliver.
Ít nhất ba đột biến gen EOGT đã được xác định ở những người mắc hội chứng Adams-Oliver. Người ta cho rằng protein EOGT điều hòa các protein Notch, từ đó kích hoạt đường tín hiệu Notch. Protein được tạo ra từ gen này biến đổi một số protein bằng cách chuyển cho chúng một phân tử N-acetylglucosamineg. Tuy nhiên, tác động của quá trình biến đổi protein đối với đường Notch và cơ chế tác động của gen EOGT gây ra bệnh vẫn chưa rõ ràng.
Chẩn đoán
Bác sĩ sẽ khám sức khỏe, kiểm tra các dấu hiệu lâm sàng cho trẻ bao gồm da đầu, dị tật bàn tay bàn chân. Thông tin tiền sử bệnh của gia đình có thể hỗ trợ quá trình chẩn đoán bệnh. Trẻ bị nghi ngờ mắc bệnh có thể thực hiện xét nghiệm di truyền tìm đột biến gen gây bệnh.
Điều trị
Hiện nay chưa có phương pháp điều trị hoàn toàn hội chứng Adams-Oliver. Các liệu pháp dựa trên triệu chứng cụ thể của mỗi trẻ mắc bệnh.
- Trẻ bị mất da đầu không thể tự lành, cần được tiến hành ghép da
- Thực hiện phẫu thuật đóng các khớp hộp sọ
- Sử dụng tay giả hoặc tập vậy lý trị liệu khắc phục dị tật ở các chi
- Khám tim, mắt định kỳ nhằm tầm soát các biến chứng và có biện pháp điều trị kịp thời
- Khám hệ thần kinh và đánh giá sự phát triển thể chất, tâm thần vận động hằng năm
Dạng di truyền
Hội chứng Adams-Oliver có hai kiểu di truyền khác nhau.
- Bệnh do các đột biến ở gen ARHGAP31, DLL4, NOTCH1 hoặc RBPJ di truyền theo kiểu trội trên nhiễm sắc thể thường. Do đó, một bản sao của gen trong mỗi tế bào đủ để gây ra bệnh. Gen đột biến thường được di truyền từ cha mẹ bị bệnh.
- Một số trường hợp liên quan đến đột biến gen NOTCH1 do các đột biến mới (de novo) xảy ra trong quá trình hình thành các tế bào sinh sản (trứng hoặc tinh trùng) hoặc trong quá trình phát triển phôi sớm. Trường hợp này xảy ra ở người không có tiền sử bệnh trong gia đình của họ.
- Bệnh do đột biến ở gen DOCK6 hoặc EOGT di truyền theo kiểu lặn trên nhiễm sắc thể thường. Do đó, bệnh chỉ biểu hiện khi có cả hai bản sao của gen đột biến trong mỗi tế bào. Bệnh nhân mắc bệnh lặn trên nhiễm sắc thể thường sẽ có bố và mẹ mang một bản sao của gen đột biến, nhưng bố mẹ ít khi biểu hiện triệu chứng bệnh.

Nguồn: U.S. National Library of Medicine
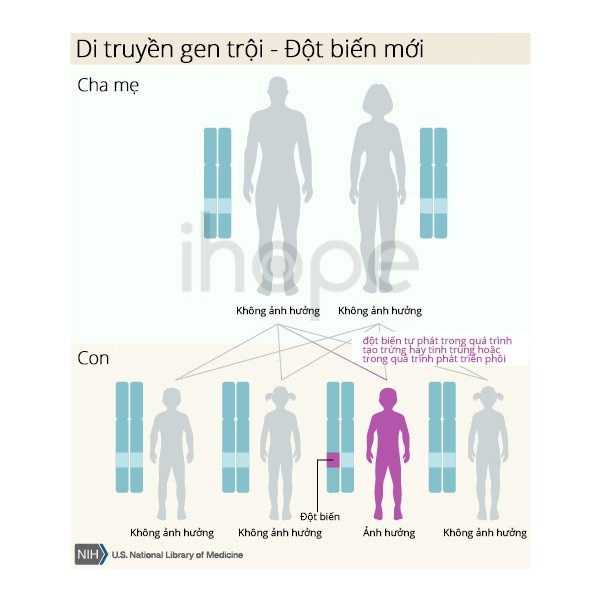
Nguồn: U.S. National Library of Medicine

Nguồn: U.S. National Library of Medicine
Phòng ngừa
Trường hợp bệnh di truyền trội trên nhiễm sắc thể thường, nếu cha hoặc mẹ mắc bệnh, con sinh ra sẽ có 50% khả năng di truyền bệnh. Do đó, để bảo đảm 100% khả năng con không bị bệnh, cha mẹ có thể chọn phương pháp thụ tinh nhân tạo IVF và sàng lọc phôi PGS/PGD.
Ngoài ra, bệnh di truyền lặn đột biến gen DOCK6 hoặc EOGT, cha mẹ mang đột biến dị hợp nên gần như không có biểu hiện bệnh, do đó rất khó phát hiện cho đến khi sinh con. Để chủ động phòng ngừa, cha mẹ nên làm xét nghiệm sàng lọc gen lặn để chủ động cho tương lai của con.
Các thành viên trong gia đình nên khám sức khỏe và tầm soát bệnh định kỳ nếu có thành viên mắc bệnh. Các cặp vợ chồng trước khi mang thai cần tư vấn và xét nghiệm di truyền đảm bảo sinh con khỏe mạnh.
Các tên gọi khác
- Absence defect of limbs, scalp, and skull
- AOS
- Aplasia cutis congenita with terminal transverse limb defects
- Congenital scalp defects with distal limb reduction anomalies
- Congenital scalp defects with distal limb reduction anomalies
- limb scalp and skull defects
- Limb, scalp, and skull defects
References
- Genetic Testing Information. Adams-Oliver syndrome. Retrieved September 16, 2022 from https://www.ncbi.nlm.nih.gov/gtr/conditions/C0265268/
- Genetic and Rare Diseases Information Center. Adams-Oliver syndrome. Retrieved September 16, 2022 from https://rarediseases.info.nih.gov/diseases/5739/adams-oliver-syndrome
- Catalog of Genes and Diseases from OMIM. ADAMS-OLIVER SYNDROME 1; AOS1. Retrieved September 16, 2022 from https://omim.org/entry/100300
- U.S National Library of Medicine. Adams-Oliver syndrome. Retrieved September 16, 2022 from https://medlineplus.gov/genetics/condition/adams-oliver-syndrome/
- National Organization for Rare Disorders. Adams-Oliver Syndrome. Retrieved September 22, 2022 from https://rarediseases.org/rare-diseases/adams-oliver-syndrome/
- Anna Lehman, MD, Wim Wuyts, PhD, and Millan S Patel, MD. Adams-Oliver Syndrome. Published April 14, 2016 https://www.ncbi.nlm.nih.gov/books/NBK355754/
- Sumara Rashid, Saleha Azeem, Samiha Riaz. Adams–Oliver Syndrome: A Rare Congenital Disorder. Published March 18, 2022 https://doi.org/10.7759/cureus.23297
- Orphanet. Adams-Oliver Syndrome. Retrieved September 22, 2022 from https://www.orpha.net/consor/cgi-bin/OC_Exp.php?lng=EN&Expert=974
- Amir Dehdashtian. Adams-Oliver syndrome: a case with full expression. Published June 27, 2016 https://doi.org/10.4081/pr.2016.6517
- MalaCards. Adams-Oliver Syndrome 5 (AOS5). Retrieved September 22, 2022 from https://www.malacards.org/card/adams_oliver_syndrome_5





