
Bệnh CLN8 (CLN8 disease) là bệnh di truyền thuộc nhóm lipofuscinoses ceroid thần kinh. Bệnh tác động chủ yếu lên hệ thần kinh với các triệu chứng liên quan đến thị lực, chuyển động và khả năng tư duy.
Nguồn: Eunice Kennedy Shriver National Institute of Child Health and Human Development
Mức độ nghiêm trọng của bệnh khác nhau đối với mỗi cá nhân. Do đó, người ta chia bệnh thành hai dạng gồm ít nghiêm trọng và nghiêm trọng.
Biểu hiện lâm sàng
Bệnh CLN8 dạng ít nghiêm trọng
Bệnh CLN8 dạng ít nghiêm trọng thường khởi phát trong giai đoạn từ 2–5 tuổi.
Bệnh bao gồm các triệu chứng phổ biến như:
- Động kinh tái phát
- Suy giảm trí tuệ
Những cơn động kinh này thuộc loại động kinh toàn thể dạng co cứng–co giật và thường không đáp ứng tốt với các liệu pháp điều trị. Khi cơn động kinh khởi phát, người bệnh xuất hiện các dấu hiệu như cứng cơ, co giật và mất ý thức. Trong một số ít trường hợp, người bệnh vẫn tỉnh táo khi cơn động kinh xảy ra. Trong độ tuổi thiếu niên, tần suất xuất hiện của các cơn động kinh khoảng 1–2 lần mỗi tháng. Khi người bệnh trưởng thành, tần suất này giảm còn 4–6 lần mỗi năm và hiếm khi xuất hiện trong độ tuổi trung niên.
Ngoài ra, bệnh nhân CLN8 còn xuất hiện các triệu chứng như suy giảm trí tuệ và chuyển động cơ thể khó khăn. Bên cạnh đó, người bệnh có thể gặp vấn đề thị giác trong giai đoạn sớm đến giữa thời kì trưởng thành. Trẻ mắc CLN8 thường không sống qua tuổi trưởng thành.
Bệnh CLN8 dạng nghiêm trọng
Bệnh CLN8 dạng nghiêm trọng thường xuất hiện trong giai đoạn từ 2–7 tuổi. CLN8 dạng này thường dẫn đến các cơn động kinh rung giật cơ.
Người bệnh thường có các biểu hiện chung như:
- Suy giảm trí tuệ
- Mất khả năng nói
- Giảm thị lực
- Vận động khó khăn và dần bất động
Sau khi các triệu chứng khởi phát, người bệnh có thể sống đến hết giai đoạn trẻ nhỏ hoặc thiếu niên.
Độ phổ biến
Bệnh CLN8 dạng ít nghiêm trọng chủ yếu ảnh hưởng đến người gốc Phần Lan, cụ thể là vùng Kainuu phía bắc Phần Lan. Tỉ lệ mắc bệnh CLN8 dạng nhẹ khoảng 1/10.000 người. Đối với dạng nghiệm trọng, tỉ lệ người bệnh chưa xác định. Nhìn chung, tất cả các dạng của bệnh NCL ảnh hưởng đến 1/100.000 người trên toàn thế giới.
Nguyên nhân
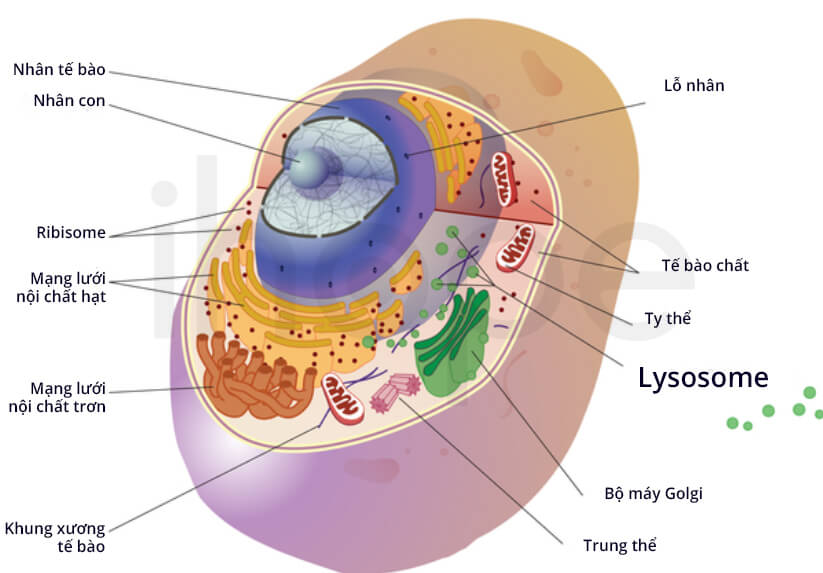
Đột biến gen CLN8 gây ra bệnh CLN8. Gen CLN8 cung cấp hướng dẫn tạo ra protein chưa rõ chức năng. Người ta cho rằng protein CLN8 giữ vai trò vận chuyển các chất qua mạng lưới nội chất. Lưới nội chất là nơi tạo ra protein, sau đó protein được đóng gói và vận chuyển đến các bào quan khác trong tế bào. Protein CLN8 hỗ trợ mạng lưới nội chất kiểm soát lượng chất béo trong tế bào. Trong một số tế bào như tế bào thần kinh, protein CLN8 hoạt động bên ngoài mạng lưới nội chất.
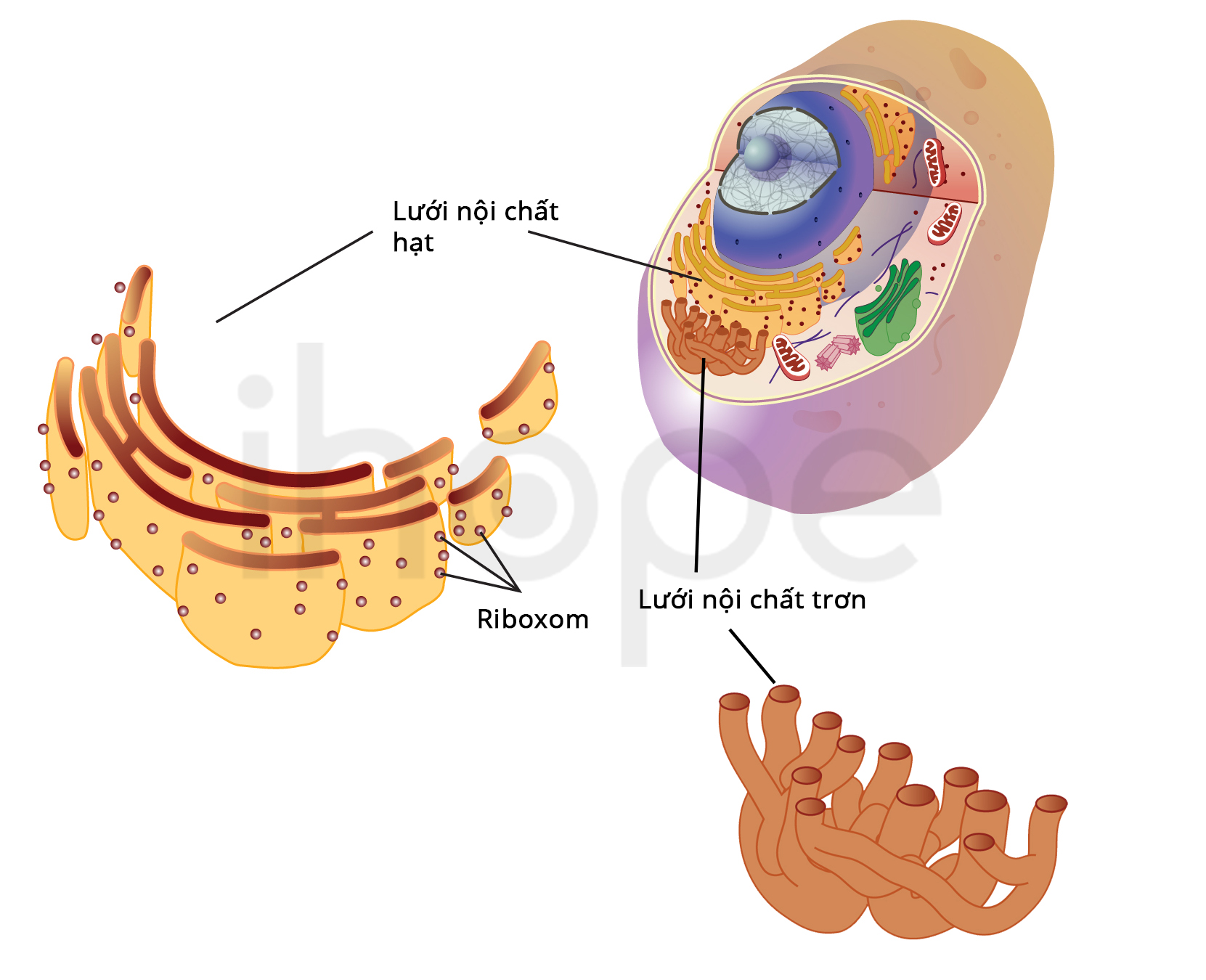
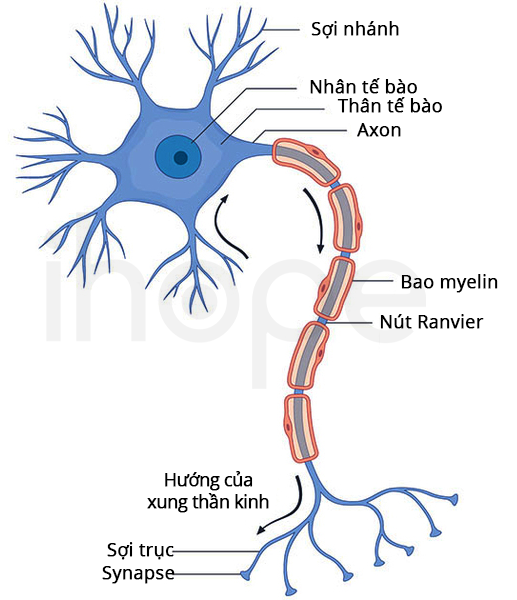
Lưới nội chất là cấu trúc bên trong tế bào tham gia vào quá trình xử lý và vận chuyển protein
Nguồn: National Human Genome Research Institute
CLN8 dạng ít nghiêm trọng
Đa số người bệnh CLN8 dạng ít nghiêm trọng đều mang đột biến gen CLN8. Tuy nhiên, trong số protein tạo ra vẫn còn một lượng ít protein bình thường. Do đó, các triệu chứng của dạng này ít nghiêm trọng hơn. Hàm lượng chất béo trong não của người bệnh giảm dần do hoạt động của protein CLN8 suy giảm. Tuy nhiên, tình trạng suy giảm chất béo trong não không gây ra hậu quả rõ ràng.
Ngoài ra, số lượng tế bào thần kinh trong não người bệnh giảm không rõ lí do gây nên nhiều bất thường về hình dạng của não. Đối với các dạng khác của bệnh, nguyên nhân dẫn đến suy giảm số lượng tế bào là do tế bào tích lũy protein và các chất khác quá mức. Ngược lại, tế bào của dạng ít nghiêm trọng chết đi do không tích lũy protein và chất khác.
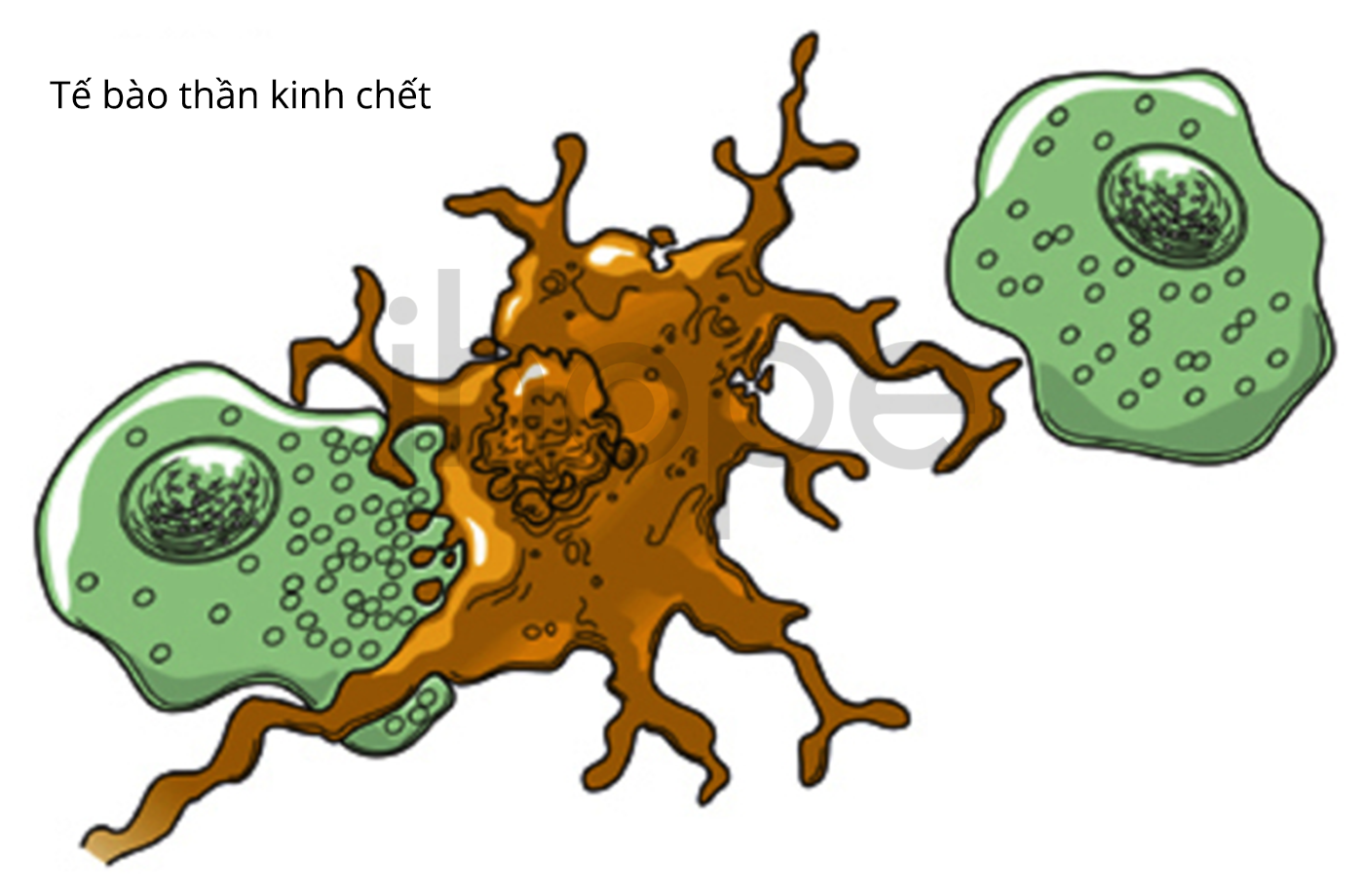
Tế bào đại thực bào (hai bên) tiêu thụ một tế bào thần kinh đang chết dần (ở giữa)
Nguồn: U.S National Library of Medicine
Đột biến gen CLN8 làm suy giảm chức năng của protein hoặc làm giảm khả năng vận chuyển protein đến mạng lưới nội chất. Do đó, chức năng của lưới nội chất suy giảm. Người ta chưa rõ tại sao số lượng protein CLN8 giảm gây nên bệnh này.
CLN8 dạng nghiêm trọng
Đối với bệnh CLN8 dạng nghiêm trọng, protein và các chất khác thường tập trung trong lysosome. Quá trình tích tụ bất thường xảy ra trong tế bào khắp cơ thể nhưng tế bào thần kinh dễ tổn thương hơn. Theo thời gian, tế bào thần kinh tổn thương và chết dần dẫn đến các dấu hiệu liên quan đến thần kinh. Trong một số trường hợp nghiêm trọng, bệnh nhân có thể tử vong. Người ta vẫn chưa xác định mối liên hệ giữa các đột biến trên gen CLN8 và quá trình tích tụ này.
Nguồn: Darryl Leja, NHGRI
Chẩn đoán
Bác sĩ chẩn đoán bệnh CLN8 thông qua dấu hiệu đặc trưng như co giật, vận động khó khăn và suy giảm trí tuệ. Ngoài ra, bác sĩ còn chỉ định thực hiện xét nghiệm máu để xác định bệnh nhân mắc bệnh NCL nhóm mấy. Người ta có thể ghi nhận cơ quan xảy ra quá trình tích trữ protein bất thường sau khi quan sát mẫu máu dưới kính hiển vi điện tử.
Ngoài ra, bệnh nhân nên thực hiện xét nghiệm di truyền để xác định chính xác đột biến gen CLN8. Người bệnh cần cung cấp mẫu máu hoặc nước bọt có chứa ADN để tiến hành xét nghiệm.
Điều trị
Hiện nay, chưa có liệu pháp điều trị hoàn toàn bệnh CLN8. Các phương pháp chủ yếu làm giảm triệu chứng và cải thiện chất lượng cuộc sống của người bệnh.
Động kinh
Bệnh nhân thường sử dụng các loại thuốc như natri valproate và clonazepam nhằm kiểm soát các cơn động kinh. Tuy nhiên, các loại thuốc này không có tác dụng làm chậm tiến triển của bệnh. Các cơn động kinh rung giật cơ có thể nghiêm trọng hơn khi người bệnh sử dụng thuốc lamotrigine.
Các biểu hiện khác
Những bệnh nhân có biểu hiện như nhai và nuốt khó khăn, tiết dịch miệng, rối loạn vận động, rối loạn giấc ngủ cần được hỗ trợ chăm sóc và điều trị bằng thuốc. Đồng thời, người bệnh nên chú ý cải thiện tư thế, khả năng vận động, chăm sóc da và miệng. Trong một số trường hợp nghiêm trọng, bác sĩ có thể chỉ định hỗ trợ bổ sung dinh dưỡng hoặc phẫu thuật đặt ống dẫn thức ăn.
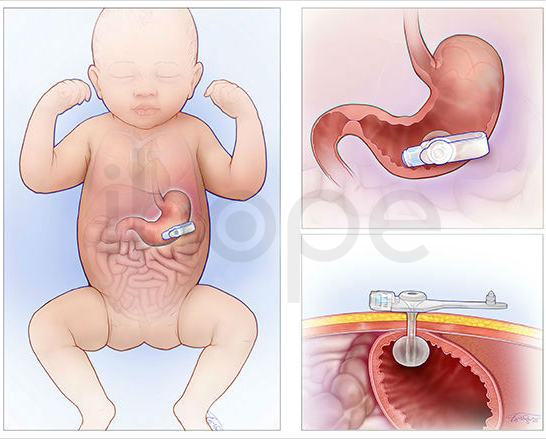
Nguồn: Children's Hospital of Phialadenphia
Dạng di truyền
Bệnh CLN8 di truyền theo kiểu lặn trên nhiễm sắc thể thường, nghĩa là cả hai bản sao của gen trong mỗi tế bào đều có đột biến. Cha mẹ của một cá nhân mắc bệnh lặn trên nhiễm sắc thể thường đều mang một bản sao của gen đột biến, nhưng họ thường không biểu hiện dấu hiệu và triệu chứng của bệnh.

Nguồn: U.S. National Library of Medicine
Phòng ngừa
Bệnh CLN8 di truyền lặn do đột biến gen CLN8, cha mẹ mang đột biến dị hợp nên gần như không có biểu hiện bệnh, do đó rất khó phát hiện cho đến khi sinh con. Cha mẹ nên làm xét nghiệm sàng lọc gen lặn để chủ động cho tương lai của con. Các cặp vợ chồng trước khi mang thai cần tư vấn và xét nghiệm di truyền nhằm đảm bảo sinh con khỏe mạnh. Người thuộc nhóm nguy cơ mắc bệnh cần khám sức khỏe và tầm soát bệnh định kỳ.
Các tên gọi khác
- Neuronal ceroid lipofuscinosis 8
References
- Genetic Testing Information. Neuronal ceroid lipofuscinosis 8 northern epilepsy variant. Retrieved June 27, 2024 from https://www.ncbi.nlm.nih.gov/gtr/conditions/C1864923/
- Genetic and Rare Diseases Information Center. Progressive epilepsy-intellectual disability syndrome, Finnish type. Retrieved June 27, 2024 from https://rarediseases.info.nih.gov/diseases/4010/index
- Catalog of Genes and Diseases from OMIM. CEROID LIPOFUSCINOSIS, NEURONAL, 8; CLN8. Retrieved June 27, 2024 from https://omim.org/entry/600143
- U.S National Library of Medicine. CLN8 disease. Retrieved June 27, 2024 from https://medlineplus.gov/genetics/condition/cln8-disease/
- National Institute of Health. CLN8 Gene Compound Heterozygous Variants: A New Case and Protein Bioinformatics Analyses. Retrieved June 27, 2024 from https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9407845/
- Oxford Academic. CLN8. Retrieved June 27, 2024 from https://academic.oup.com/book/30048/chapter-abstract/256054781
- Orphanet. CLN8 disease. Retrieved June 27, 2024 from https://www.orpha.net/en/disease/detail/228354
- Frontiers. Two compound heterozygous variants in the CLN8 gene are responsible for neuronal cereidolipofuscinoses disorder in a child: a case report. Retrieved June 27, 2024 from https://www.frontiersin.org/journals/pediatrics/articles/10.3389/fped.2024.1379254/full





