
Bệnh thiếu cofactor molybdenum (molybdenum cofactor deficiency) hiếm gặp và thường gây tổn hại chức năng não. Người ta chia bệnh thành hai dạng là thiếu cofactor molybdenum khởi phát sớm trong giai đoạn sơ sinh. Dạng thứ hai là thiếu cofactor molybdenum khởi phát muộn từ giai đoạn trẻ nhỏ đến trưởng thành.
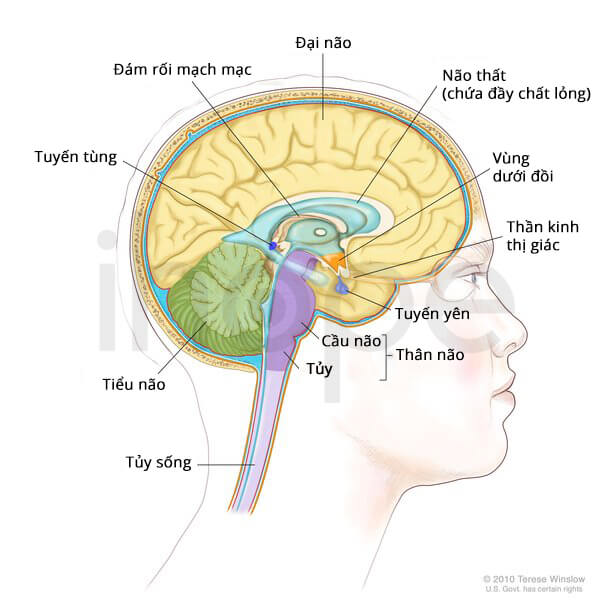
Nguồn: Terese Winslow
Biểu hiện lâm sàng
Thiếu cofactor molybdenum khởi phát sớm
Thiếu cofactor molybdenum khởi phát sớm thuộc dạng nặng. Bệnh nhi thường xuất hiện các triệu chứng bệnh liên quan đến não ngay khi mới sinh. Động kinh là dấu hiệu phổ biến nhất. Trẻ thường có biểu hiện động kinh trong hai tuần sau sinh. Một số biểu hiện co giật khác bao gồm rung giật nhãn cầu và co giật khi ngủ.
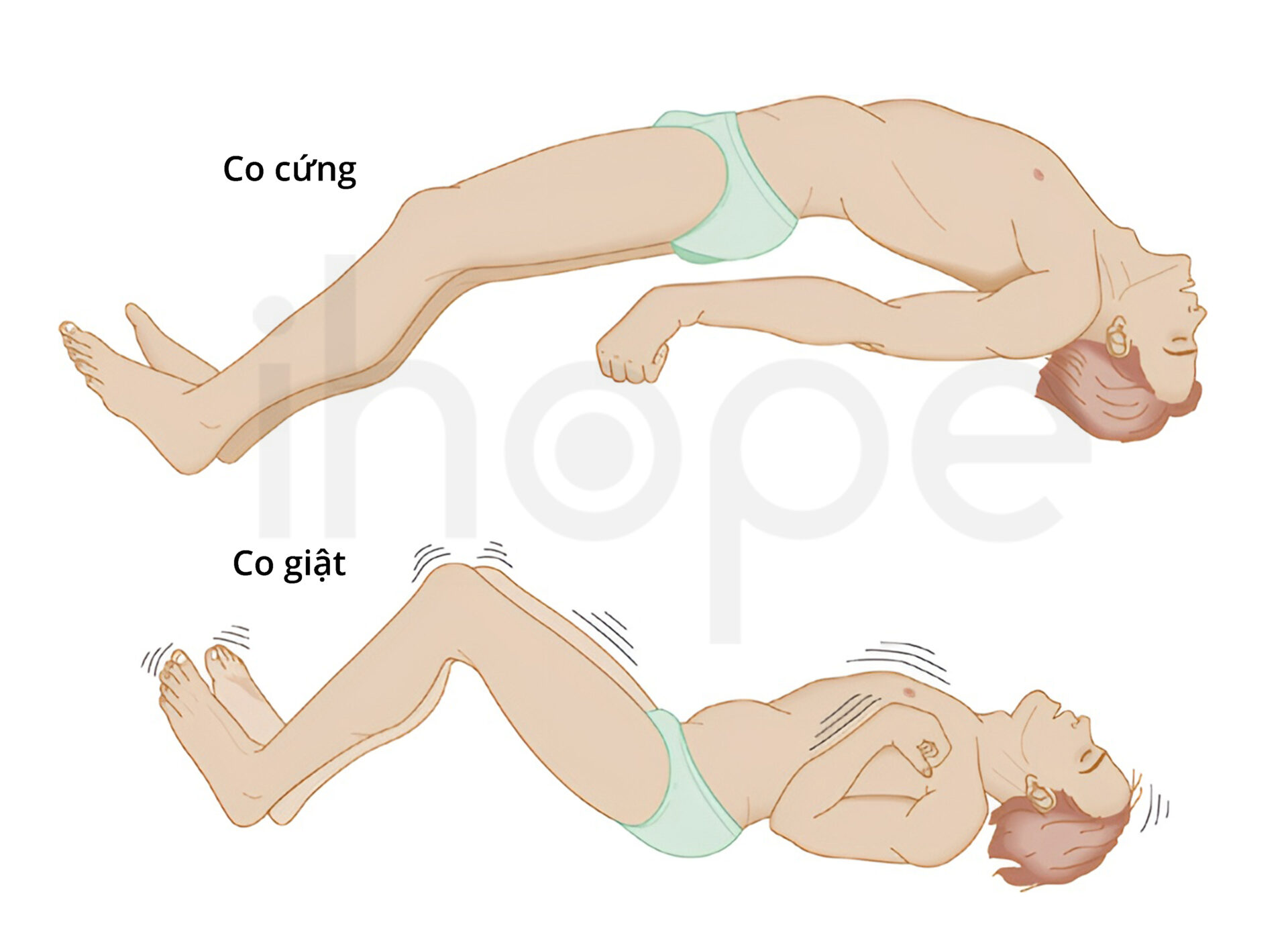
Nguồn: NEUROSURGERY Neurosurgery.
Trẻ có phản xạ quá mức đối với các kích thích âm thanh, hình ảnh hoặc tiếp xúc trên cơ thể. Những phản xạ này có thể là giật mình mạnh hoặc co cứng người. Người ta cũng ghi nhận một số bệnh nhân có các bệnh lý về mắt nhưng đây không phải là dấu hiệu phổ biến. Trẻ mới sinh có kích thước đầu bình thường. Tuy nhiên, quá trình thoái hoá não diễn ra dẫn đến kích thước đầu nhỏ khi trẻ trưởng thành.
Nguồn: National Human Genome Research Institute
Bệnh nhân có những điểm dị biệt trên khuôn mặt bao gồm:
- Thái dương hẹp
- Dáng mặt dài
- Gò má lồi
- Mắt lõm, hai mắt cách xa nhau
- Khe mí mắt dài
- Nhân trung dài
- Mũi nhỏ
- Môi dày
Ảnh: Trán cao thu hẹp lại ở thái dương
Nguồn: Elements of Morphology, National Human Genome Research Institute
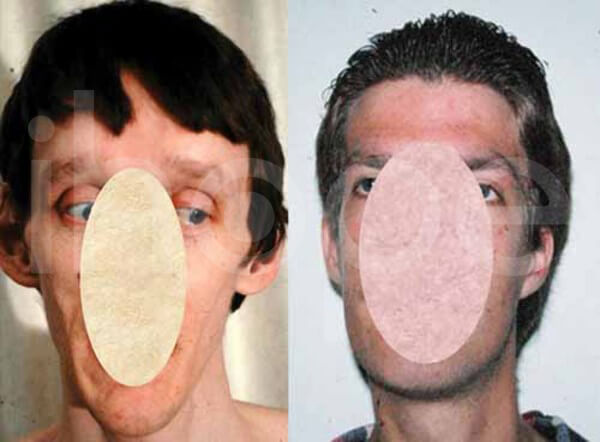
Ảnh: Khuôn mặt dài
Nguồn: National Human Genome Research Institute
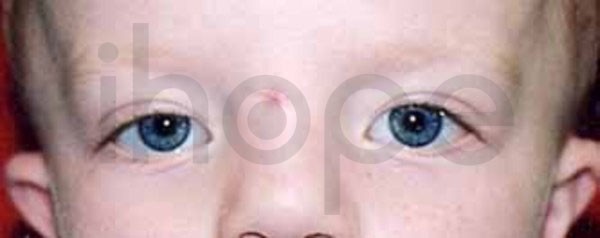
Ảnh: Khoảng cách giữa hai mắt rộng
Nguồn: Elements of Morphology, National Human Genome Research Institute
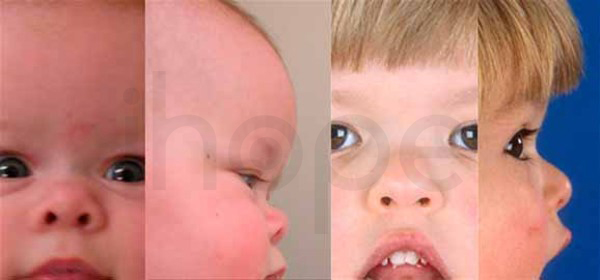
Ảnh: Mũi nhỏ
Nguồn: Elements of Morphology, National Human Genome Research Institute
Bên cạnh đó, trẻ thiếu cofactor molybdenum có thể gặp một số vấn đề như:
- Suy giảm khả năng ngôn ngữ
- Vấn đề hô hấp như ngừng thở khi ngủ và viêm phổi hít
- Khó nuốt và dễ sặc
Thiếu cofactor molybdenum khởi phát muộn
Thiếu cofactor molybdenum khởi phát muộn là dạng nhẹ. Bệnh nhân xuất hiện một số triệu chứng tương tự dạng khởi phát sớm nhưng ít nghiêm trọng hơn.
Người bệnh có các dấu hiệu suy giảm chức năng thần kinh-vận động bao gồm:
- Tinh thần không ổn định
- Loạn trương lực cơ
- Co giật không tự chủ
- Mất khả năng phối hợp cử động
Bên cạnh đó, người bệnh có triệu chứng khó ăn và bị lệch thuỷ tinh thể. Tuy nhiên, khác với dạng khởi phát sớm, họ có khả năng nhận thức bình thường và chỉ chậm phát triển mức độ nhẹ. Triệu chứng động kinh ít phổ biến đối với bệnh nhân thiếu cofactor molybdenum khởi phát muộn.
Nguồn: ARZTSAMUI/Shutterstock.com
Độ phổ biến
Thiếu cofactor molybdenum rất hiếm gặp. Theo ước tính, trên toàn thế giới có 1/100.000–200.000 trẻ sơ sinh mắc bệnh. Người ta đã ghi nhận hơn 100 ca bệnh nhưng số liệu thực tế có thể nhiều hơn.
Nguyên nhân
Đột biến gen MOCS1, MOCS2 và GPHN lần lượt gây ra bệnh thiếu cofactor molybdenum. Gen MOCS1 cung cấp hướng dẫn tạo ra protein MOCS1A và MOCS1B. Gen MOCS2 cung cấp hướng dẫn tạo ra protein MOCS2A và MOCS2B. Bốn loại protein này kết hợp với nhau tạo thành phức hợp protein molybdopterin synthase. Gen GPHN cung cấp hướng dẫn tạo ra protein gephyrin. Ngoài ra, người ta cũng ghi nhận một trường hợp bệnh liên quan đến đột biến gen MOCS3. Gen này cung cấp hướng dẫn tạo ra protein MOCS3. Các protein này đều tham gia quá trình tổng hợp cofactor molybdenum.
Cofactor molypden có chứa nguyên tố molypden rất quan trọng đối với khả năng hoạt động của một số enzyme như sulfite oxidase, idehyde oxidase và xanthine dehydrogenase. Những enzyme này có chức năng phân giải hoặc chuyển hóa nhiều chất khác nhau trong cơ thể, trong đó một số chất sẽ gây hại nếu không được chuyển hóa.
Đột biến các gen MOCS1, MOCS2 hoặc GPHN làm suy giảm hoặc mất chức năng của những protein tham gia quá trình tổng hợp cofactor molybdenum. Khi đó, enzyme không có cofactor molybdenum nên nó không thể thực hiện chức năng phân giải hoặc chuyển hóa các chất. Vì vậy, một số chất như sulfite, S-sulfocysteine, xanthine tích tụ trong cơ thể đến mức gây độc.
Trong các chất trên, sulfite có thể gây độc cho não. Nồng độ sulfite cao bất thường có thể dẫn dến các bệnh lý về não, động kinh và những triệu chứng khác của bệnh thiếu cofactor molybdenum.
Chẩn đoán
Bác sĩ thường chẩn đoán thiếu cofactor molybdenum dựa trên các biểu hiện lâm sàng, xét nghiệm máu, nước tiểu và xét nghiệm di truyền.
Xét nghiệm máu
Kết quả xét nghiệm máu của người bệnh sẽ có chỉ số:
- Taurine tăng
- Cystine giảm
- Homocysteine giảm
Xét nghiệm nước tiểu
Kết quả xét nghiệm nước tiểu của người bệnh sẽ có chỉ số:
- Xanthine tăng
- Hypoxanthine tăng
- Acid uric giảm
- S-sulfocysteine tăng
- Thiosulfate tăng
Xét nghiệm di truyền
Bệnh nhân cần thực hiện xét nghiệm di truyền nếu kết quả xét nghiệm máu hoặc nước tiểu cho thấy có dấu hiệu thiếu cofactor molybdenum. Bác sĩ có thể chỉ định xét nghiệm các gen GPHN, MOCS1, MOCS2 và MOCS3 để tìm đột biến gây bệnh.
Điều trị
Bác sĩ thường kê đơn fosdenopterin dạng uống hoặc tiêm cho bệnh nhân thiếu cofactor molybdenum nhằm khôi phục quá trình tổng hợp cofactor molybdenum trong cơ thể. Ngoài ra, bệnh nhân có thể áp dụng chế độ ăn ít protein để giảm quá trình tổng hợp sulfite.
Các phương pháp giúp bệnh nhân cải thiện triệu chứng bệnh bao gồm:
- Sử dụng thuốc chống động kinh
- Đặt ống thông dạ dày
- Bổ sung thiamine
- Tiêm botulinum toxin hoặc phenol
- Bổ sung magnesium
- Phẫu thuật mở khí quản
Dạng di truyền
Thiếu cofactor molybdenum di truyền theo kiểu lặn trên nhiễm sắc thể thường. Do đó, bệnh chỉ biểu hiện khi có cả hai bản sao của gen đột biến trong mỗi tế bào. Bệnh nhân mắc bệnh lặn trên nhiễm sắc thể thường sẽ có bố và mẹ mang một bản sao của gen đột biến, nhưng họ ít khi biểu hiện triệu chứng bệnh.

Nguồn: U.S. National Library of Medicine
Người ta cũng ghi nhận ít nhất một bệnh nhân thiếu cofactor molybdenum do nhận hai bản sao gen MOCS1 đột biến từ bố hoặc mẹ thay vì một bản sao như thông thường. Đột biến này phát sinh trong quá trình hình thành tế bào trứng hoặc tinh trùng.
Phòng ngừa
Bệnh di truyền lặn do đột biến nhiều gen, cha mẹ mang đột biến dị hợp nên gần như không có biểu hiện bệnh, do đó rất khó phát hiện cho đến khi sinh con. Để chủ động phòng ngừa, cha mẹ nên làm xét nghiệm sàng lọc gen lặn để chủ động cho tương lai của con. Các cặp vợ chồng trước khi mang thai cần tư vấn và xét nghiệm di truyền nhằm đảm bảo sinh con khỏe mạnh. Người thuộc nhóm nguy cơ mắc bệnh cần khám sức khỏe và tầm soát bệnh định kỳ.
Các tên gọi khác
- MOCOD
- Deficiency of molybdenum cofactor
- Combined molybdoflavoprotein enzyme deficiency
- Combined xanthine oxidase and sulfite oxidase and aldehyde oxidase deficiency
- Combined deficiency of sulfite oxidase, xanthine dehydrogenase, and aldehyde oxidase
References
- Genetic Testing Information. Combined molybdoflavoprotein enzyme deficiency. Retrieved June 04, 2024 from https://www.ncbi.nlm.nih.gov/gtr/conditions/C0268119/
- Genetic and Rare Diseases Information Center. Sulfite oxidase deficiency due to molybdenum cofactor deficiency. Retrieved June 04, 2024 from https://rarediseases.info.nih.gov/diseases/3705/index
- Catalog of Genes and Diseases from OMIM. MOLYBDENUM COFACTOR DEFICIENCY, TYPE A; MOCODA. Retrieved June 04, 2024 from https://omim.org/entry/252150
- U.S National Library of Medicine. Molybdenum cofactor deficiency. Retrieved June 04, 2024 from https://medlineplus.gov/genetics/condition/molybdenum-cofactor-deficiency/
- MalaCards. Molybdenum Cofactor Deficiency (MOCOD). Retrieved June 04, 2024 from https://www.malacards.org/card/molybdenum_cofactor_deficiency
- National Institute of Health. Molybdenum Cofactor Deficiency. Retrieved June 04, 2024 from https://www.ncbi.nlm.nih.gov/books/NBK575630/
- National Organization for Rare Disorders. Molybdenum Cofactor Deficiency. Retrieved June 04, 2024 from https://rarediseases.org/rare-diseases/molybdenum-cofactor-deficiency-mocd/
- Orphanet. Sulfite oxidase deficiency due to molybdenum cofactor deficiency. Retrieved June 04, 2024 from https://www.orpha.net/en/disease/detail/99732





