Alpha-mannosidosis là một bệnh di truyền hiếm gặp do cơ thể không có khả năng phân hủy một số nhóm đường phức tạp trong tế bào. Những phân tử này tích tụ gây ảnh hưởng đến nhiều mô và cơ quan (bao gồm cả hệ thần kinh trung ương), từ đó gây ra các triệu chứng như thiểu năng trí tuệ, đặc điểm dị biệt trên khuôn mặt và các bất thường về xương. Mức độ nghiêm trọng của triệu chứng khác nhau đáng kể giữa các bệnh nhân, trong khi một số trẻ mắc bệnh nặng và tiến triển nhanh chóng, những trẻ khác chỉ xuất hiện một ít triệu chứng nhẹ. Đây là 1 trong khoảng 50 bệnh thuộc nhóm bệnh lưu trữ lysosome (LSD). Các đặc điểm dị biệt trên khuôn mặt có thể bao gồm đầu to, trán dô, đường chân tóc thấp, lông mày cong, tai to, sống mũi dẹt, hàm nhô ra, răng thưa, nướu mọc quá mức và lưỡi to. Bệnh cũng có thể gây ra các bất thường về xương như loãng xương, đỉnh hộp sọ dày lên, biến dạng xương cột sống, vẹo ngoài khớp gối và thoái hóa xương khớp.
Ảnh: Dị tật đầu to Ảnh: Trán nhô ra phía trước Ảnh: Đường chân tóc thấp Ảnh: Hàm dưới nhô ra Alpha-mannosidosis có thể tiến triển thành các bệnh bao gồm:
Ảnh: Đục thủy tinh thể Trong một số trường hợp, bệnh nhân có thể gặp phải các triệu chứng tâm thần như trầm cảm, lo lắng hoặc ảo giác. Người ta cho rằng nguyên nhân gây ra rối loạn tâm thần có thể do các yếu tố gây căng thẳng như phẫu thuật, cảm xúc khó chịu hoặc thay đổi thói quen. Triệu chứng của alpha-mannosidosis khác nhau giữa từng bệnh nhân với mức độ từ nhẹ đến nặng. Bệnh có thể xuất hiện trong giai đoạn sơ sinh với tốc độ tiến triển nhanh chóng và gây suy thoái thần kinh nghiêm trọng. Trẻ mắc bệnh alpha-mannosidosis khởi phát sớm thường không thể sống sót qua giai đoạn trẻ nhỏ. Đối với những trường hợp nghiêm trọng, thai nhi có thể chết trước khi sinh. Người khác mắc bệnh alpha-mannosidosis có triệu chứng nhẹ, xuất hiện muộn, tiến triển chậm hơn; họ có thể sống đến 50 tuổi. Người ta ước tính tỉ lệ mắc bệnh alpha-mannosidosis khoảng 1/500.000 người trên toàn thế giới. Đột biến gen MAN2B1 gây ra bệnh alpha-mannosidosis. Gen MAN2B1 cung cấp hướng dẫn tạo ra enzyme alpha-mannosidase hoạt động trong lysosome—bào quan có vai trò tiêu hóa và tái chế vật liệu trong tế bào. Lysosome chứa các enzyme có khả năng phân hủy phức hợp đường (oligosaccharide) gắn với protein (glycoprotein). Trong đó, enzyme alpha-mannosidase tham gia vào quá trình phá vỡ các phức hợp phân tử đường chứa mannose. Đột biến gen MAN2B1 làm cho enzyme alpha-mannosidase suy yếu chức năng, từ đó chúng ngăn cản enzyme phá vỡ các oligosaccharide chứa mannose. Những oligosaccharide này tích tụ nhiều trong lysosome khiến cho các tế bào hoạt động bất thường, cuối cùng dẫn đến chết tế bào. Các mô và cơ quan bị tổn thương, từ đó triệu chứng của bệnh alpha-mannosidosis khởi phát. Bác sĩ chẩn đoán bệnh alpha-mannosidosis bằng cách nhận diện các triệu chứng, đánh giá lâm sàng chi tiết, xem xét tiền sử bệnh nhân và thực hiện các xét nghiệm chuyên biệt. Trong đó, xét nghiệm chuyên biệt có thể phát hiện mức độ thiếu hụt hoặc khả năng hoạt động của enzyme alpha-mannosidase trong tế bào bạch cầu hoặc nguyên bào sợi nuôi cấy. Ngoài ra, bệnh nhân có thể thực hiện xét nghiệm di truyền nhằm xác nhận kết quả chẩn đoán. Phân tích nước tiểu có thể phát hiện một số oligosacarit giàu mannose có nồng độ cao. Tuy nhiên, phương pháp này chỉ cho thấy nguy cơ mắc bệnh và không có giá trị chẩn đoán. Vào năm 2023, Cơ quan Quản lí Thực phẩm và Dược phẩm Hoa Kì (FDA) đã phê duyệt velmanase alfa (Lamzede) là liệu pháp thay thế enzyme đầu tiên trong điều trị các triệu chứng không liên quan đến thần kinh. Ngoài ra, người ta còn sử dụng một số phương pháp khác nhằm giảm nhẹ triệu chứng và cải thiện cuộc sống cho người bệnh. Bệnh nhân có thể sử dụng thuốc kháng sinh khi gặp nhiễm trùng do vi khuẩn hoặc dùng máy trợ thính, ống cân bằng áp suất khi mắc các vấn đề về thính giác. Bác sĩ cũng có thể khuyến nghị điều trị vật lí trị liệu cho những bệnh nhân yếu cơ. Các biện pháp can thiệp chỉnh hình bao gồm phẫu thuật hoặc sử dụng các thiết bị hỗ trợ có thể cần thiết trong điều trị các bất thường liên quan đến xương. Trong một số trường hợp, người bệnh có thể cần sử dụng xe lăn. Bệnh nhân mắc não úng thủy có thể được điều trị bằng cách đặt một ống (shunt) nhằm dẫn lưu dịch não tủy dư thừa ra khỏi não. Trẻ mắc alpha-mannosidosis cần được can thiệp càng sớm càng tốt nhằm giảm thiểu tác hại của bệnh. Các dịch vụ có thể cần thiết cho trẻ mắc bệnh bao gồm giáo dục đặc biệt, trị liệu ngôn ngữ, dịch vụ chuyên biệt dành cho trẻ khiếm thính và các dịch vụ y tế, xã hội hoặc nghề nghiệp. Ngoài ra, các chuyên gia khuyến khích gia đình và bệnh nhân nên thăm khám tư vấn di truyền . Alpha-mannosidosis di truyền theo kiểu lặn trên nhiễm sắc thể thường. Do đó, cả hai bản sao của gen trong mỗi tế bào đều mang đột biến. Bố mẹ của một người mắc bệnh thường mang một bản sao của gen đột biến nhưng không biểu hiện các dấu hiệu và triệu chứng của bệnh. Alpha-mannosidosis di truyền lặn do đột biến gen MAN2B1, cha mẹ mang đột biến dị hợp nên gần như không có biểu hiện bệnh, do đó rất khó phát hiện cho đến khi sinh con. Cha mẹ nên làm xét nghiệm sàng lọc gen lặn để chủ động cho tương lai của con. Các cặp vợ chồng trước khi mang thai cần tư vấn và xét nghiệm di truyền nhằm đảm bảo sinh con khỏe mạnh. Người thuộc nhóm nguy cơ mắc bệnh cần khám sức khỏe và tầm soát bệnh định kỳ.Biểu hiện lâm sàng
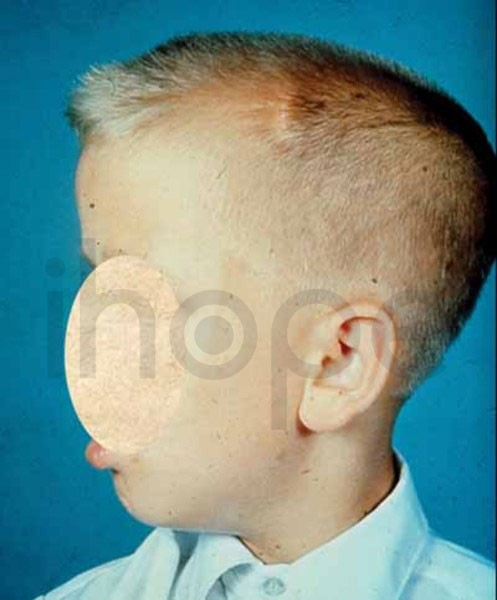
Nguồn: National Library of Medicine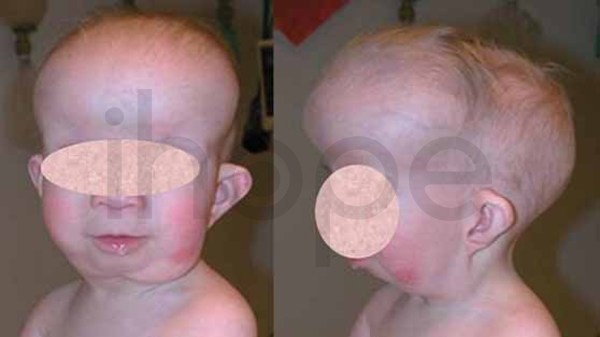
Nguồn: Elements of Morphology, National Human Genome Research Institute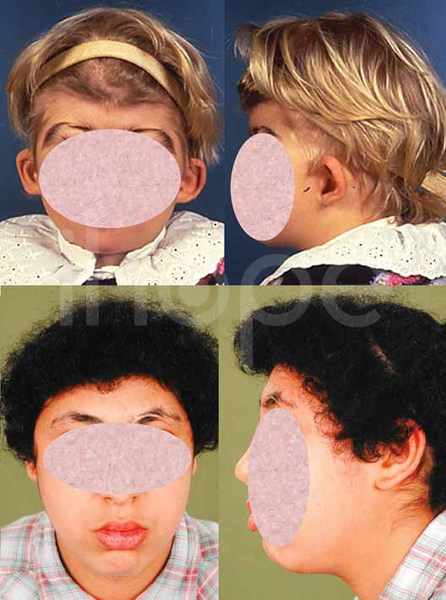
Nguồn: Elements of Morphology, National Human Genome Research Institute.
Nguồn: Elements of Morphology, National Human Genome Research Institute.
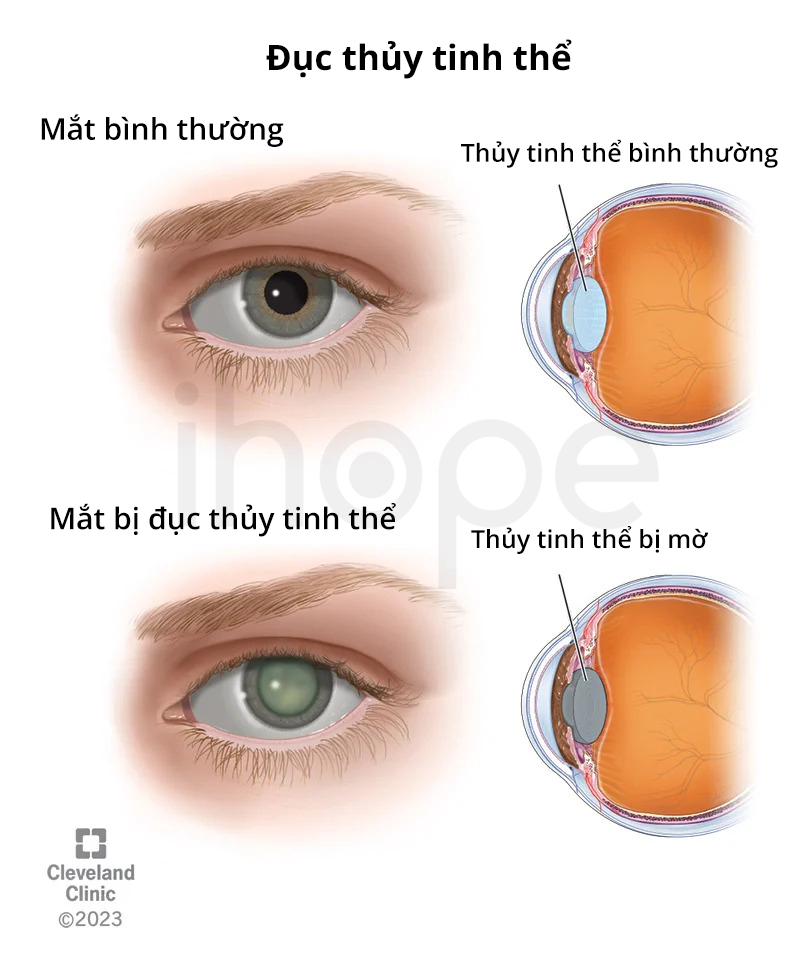
Nguồn: Cleveland ClinicĐộ phổ biến
Nguyên nhân
Chẩn đoán
Điều trị
Dạng di truyền

Nguồn: U.S. National Library of MedicinePhòng ngừa
Các tên gọi khác
References
- Genetic Testing Information. Deficiency of alpha-mannosidase. Retrieved February 06, 2024 from https://www.ncbi.nlm.nih.gov/gtr/conditions/C0024748/
- Genetic and Rare Diseases Information Center. Alpha-mannosidosis. Retrieved February 06, 2024 from https://rarediseases.info.nih.gov/diseases/6968/index
- Catalog of Genes and Diseases from OMIM. MANNOSIDOSIS, ALPHA B, LYSOSOMAL; MANSA. Retrieved February 06, 2024 from https://omim.org/entry/248500
- MedlinePlus. Alpha-mannosidosis. Retrieved February 06, 2024 from https://medlineplus.gov/genetics/condition/alpha-mannosidosis/
- National Organization for Rare Disorders. Alpha-Mannosidosis. Retrieved February 06, 2024 from https://rarediseases.org/rare-diseases/alpha-mannosidosis/