Bệnh lưu trữ chylomicron là bệnh di truyền làm suy yếu quá trình hấp thu chất béo, cholesterol và một số vitamin từ thực phẩm. Bệnh chủ yếu ảnh hưởng đến hệ tiêu hóa và hệ thần kinh.
Biểu hiện lâm sàng
Bệnh lưu trữ chylomicron khởi phát ngay sau sinh hoặc giai đoạn trẻ nhỏ. Trẻ mắc bệnh thường có những biểu hiện bao gồm:
- Chậm tăng trưởng và phát triển dẫn đến nhẹ cân
- Tiêu chảy thường xuyên (mãn tính)
- Phân có mùi hôi (phân mỡ, phân có dầu)
- Nồng độ cholesterol trong máu giảm (hypocholesterolemia)
Một số trường hợp mắc bệnh phát triển chứng tích tụ chất béo bất thường trong gan (gan nhiễm mỡ) dẫn đến gan phình to.
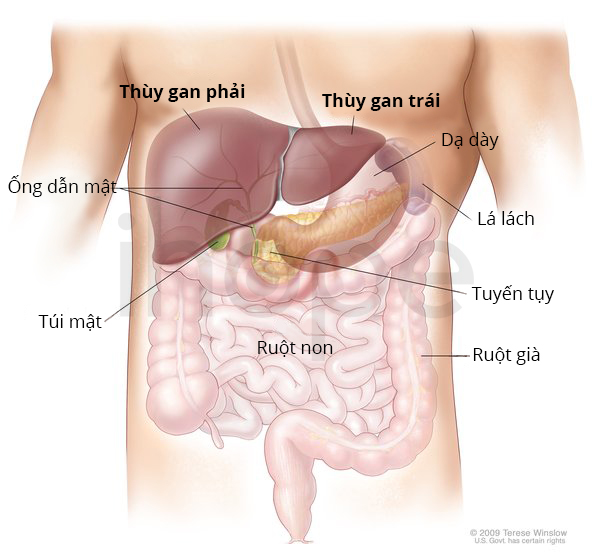
Nguồn: Medlineplus.gov
Những biểu hiện khác của bệnh lưu giữ chylomicron có thể khởi phát muộn trong giai đoạn trẻ nhỏ và thường làm giảm chức năng hệ thần kinh. Bệnh nhân gặp một số vấn đề như giảm phản xạ (hyporeflexia) và giảm khả năng cảm nhận rung động. Ngoài ra, họ cũng có thể bị bất thường về tim hoặc teo cơ (amyotrophy) nhưng thường hiếm gặp.
Độ phổ biến
Bệnh lưu trữ chylomicron hiếm gặp. Khoảng 50 trường hợp mắc bệnh đã được ghi nhận trên toàn thế giới.
Nguyên nhân
Đột biến gen SAR1B gây ra bệnh lưu trữ chylomicron. Gen SAR1B cung cấp hướng dẫn tạo ra protein cần thiết cho quá trình vận chuyển các phân tử chylomicron. Trong giai đoạn tiêu hóa, chylomicron hình thành trong các tế bào enterocyte. Những tế bào này hiện diện trong ruột non nhằm hấp thụ các chất dinh dưỡng. Cơ thể cần chylomicron để hấp thụ các vitamin tan trong dầu và vận chuyển chất béo, cholesterol từ ruột non vào máu.
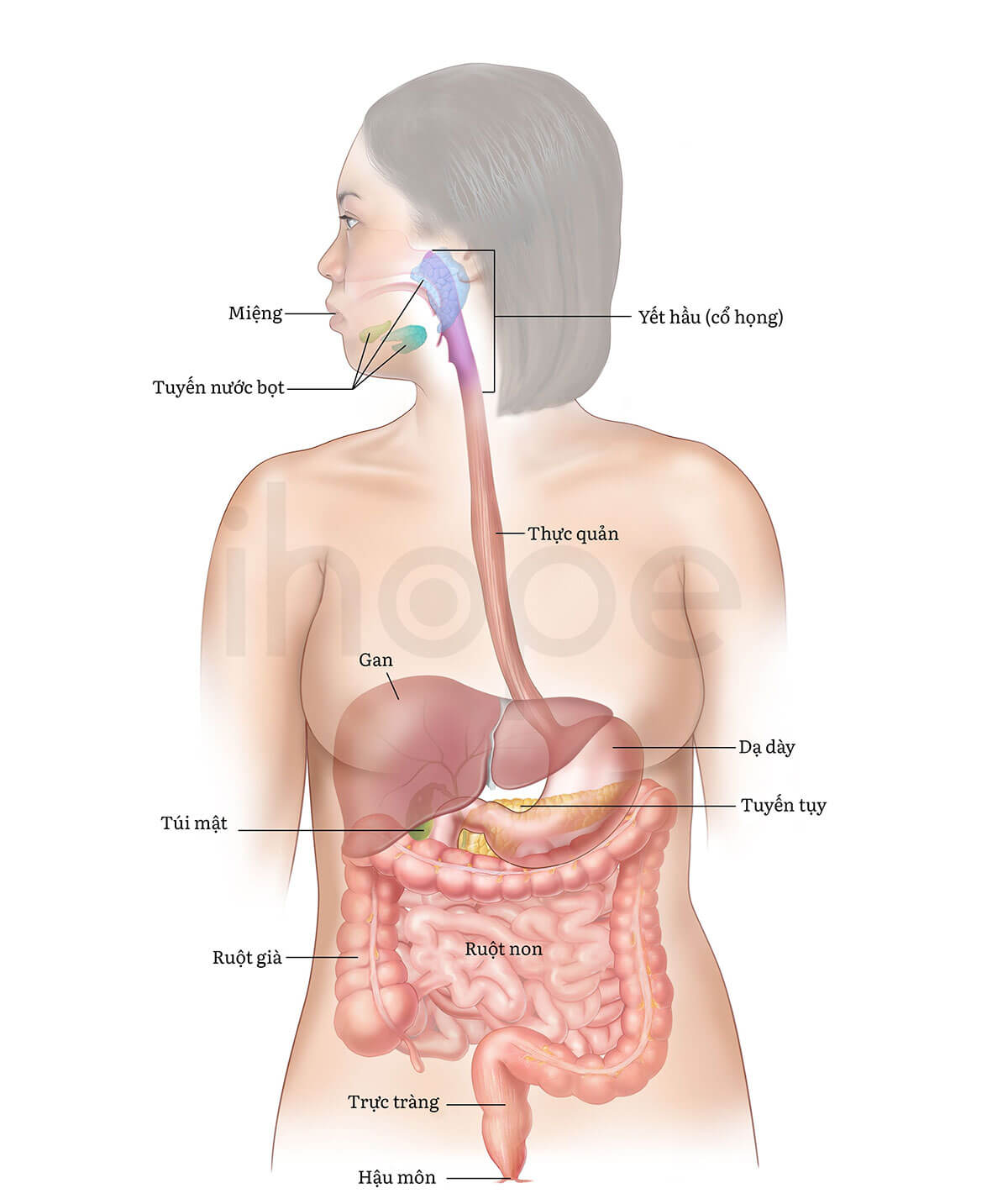
Ảnh: Hệ tiêu hóa
Nguồn: National Cancer
Đột biến gen SAR1B khiến chylomicron bị giữ trong tế bào enterocyte và không thể đi vào máu, dẫn đến suy giảm nghiêm trọng hiệu quả hấp thu chất béo và vitamin tan trong dầu từ thực phẩm. Người bệnh không thể hấp thu đủ chất béo, cholesterol và vitamin cần thiết để tăng trưởng và phát triển.
Chẩn đoán
Các biểu hiện nghi ngờ mắc bệnh lưu trữ chylomicron bao gồm chậm phát triển, tiêu chảy mãn tính và phân có mỡ. Bác sĩ sẽ chỉ định một số xét nghiệm máu chuyên biệt. Kết quả xét nghiệm cho thấy nồng độ cholesterol trong máu, apolipoprotein B và chất béo trung tính trong huyết tương thấp. Các vitamin tan trong dầu như vitamin A, D, E giảm và mức Creatine kinase huyết tương tăng cao. Xét nghiệm di truyền phân tử nhằm phát hiện các đột biến gen liên quan cũng có thể được chỉ định.
Điều trị
Hiện nay chưa có phương pháp điều trị dứt điểm bệnh lưu trữ chylomicron. Các liệu pháp giúp giảm nhẹ những triệu chứng và cải thiện chất lượng đời sống bệnh nhân. Người bệnh cần nạp đủ lượng calo bằng chế độ ăn ít chất béo (<30% tổng lượng calo từ chất béo). Tuy nhiên, họ vẫn phải bổ sung đủ các axit béo thiết yếu, có thể có hoặc không chất béo trung tính chuỗi trung bình (medium-chain triglycerides). Ngoài ra, bác sĩ sẽ chỉ định vitamin tan trong dầu liều cao, bao gồm vitamin E, A, D với liều lượng phù hợp. Trường hợp mắc bệnh chẩn đoán muộn dẫn đến các biến chứng thần kinh, bổ sung vitamin qua đường tĩnh mạch có thể được cân nhắc dù hiệu quả điều trị vẫn chưa rõ ràng. Bác sĩ sẽ chỉ định phác đồ điều trị tiêu chuẩn đối với các khiếm khuyết về thị lực, mất điều hòa vận động và bệnh cơ tim.
Dạng di truyền
Bệnh lưu trữ chylomicron di truyền theo kiểu lặn trên nhiễm sắc thể thường. Do đó, bệnh chỉ biểu hiện khi có cả hai bản sao của gen đột biến trong mỗi tế bào. Bệnh nhân mắc bệnh lặn trên nhiễm sắc thể thường sẽ có bố và mẹ mang một bản sao của gen đột biến, nhưng bố mẹ ít khi biểu hiện triệu chứng bệnh.

Nguồn: U.S. National Library of Medicine
Phòng ngừa
Bệnh lưu trữ chylomicron di truyền lặn đột biến gen SAR1B, cha mẹ mang đột biến dị hợp nên gần như không có biểu hiện bệnh, do đó rất khó phát hiện cho đến khi sinh con. Để chủ động phòng ngừa, cha mẹ nên làm xét nghiệm sàng lọc gen lặn để chủ động cho tương lai của con. Các cặp vợ chồng trước khi mang thai cần tư vấn và xét nghiệm di truyền đảm bảo sinh con khỏe mạnh. Người thuộc nhóm nguy cơ mắc bệnh cần khám sức khỏe và tầm soát bệnh định kỳ.
Các tên gọi khác
- Anderson disease
- Anderson syndrome
- CMRD
- Hypobetalipoproteinemia with accumulation of apolipoprotein B-like protein in intestinal cells
- Lipid transport defect of intestine
References
- Genetic Testing Information. Chylomicron retention disease. Retrieved 25 May 2023 from https://www.ncbi.nlm.nih.gov/gtr/conditions/C0795956/?_ga=2.49166305.63969999.1684744604-1635318420.1684225451
- Genetic and Rare Diseases Information Center. Chylomicron retention disease. Retrieved 25 May 2023 from https://rarediseases.info.nih.gov/diseases/9683/chylomicron-retention-disease
- Catalog of Genes and Diseases from OMIM. CHYLOMICRON RETENTION DISEASE; CMRD. Retrieved 25 May 2023 from https://omim.org/entry/246700
- U.S National Library of Medicine. Chylomicron retention disease Retrieved 25 May 2023 from https://medlineplus.gov/genetics/condition/chylomicron-retention-disease/
- Frontiers. From Congenital Disorders of Fat Malabsorption to Understanding Intra-Enterocyte Mechanisms Behind. Chylomicron Assembly and Secretion Retrieved 25 May 2023 from https://www.frontiersin.org/articles/10.3389/fphys.2021.629222/full
- MalaCards. Chylomicron Retention Disease (CMRD) Retrieved 25 May 2023 from https://www.malacards.org/card/chylomicron_retention_disease
- National Organization for Rare Disorders. Chylomicron retention disease Retrieved 25 May 2023 from https://rarediseases.org/gard-rare-disease/chylomicron-retention-disease/
- Orphanet. Chylomicron retention disease. Retrieved 25 May 2023 from https://www.orpha.net/consor/cgi-bin/OC_Exp.php?Expert=71&lng=EN
- Radiopaedia. Low phospholipid-associated cholelithiasis syndrome. Retrieved 25 May 2023 from https://radiopaedia.org/articles/low-phospholipid-associated-cholelithiasis-syndrome