
Hội chứng Alport là bệnh di truyền hiếm gặp đặc trưng bởi bệnh thận, suy giảm thính lực và các bệnh về mắt. Bệnh di truyền theo kiểu liên kết với nhiễm sắc thể giới tính X phổ biến nhất. Trong trường hợp này, nam giới mắc bệnh thường có biểu hiện bệnh nặng hơn nữ giới mắc bệnh.
Biểu hiện lâm sàng
Hầu hết những người mắc bệnh Alport đều bị suy giảm chức năng thận. Suy giảm chức năng trong thời gian dài làm tích tụ chất cặn bã trong cơ thể dẫn đến bệnh thận giai đoạn cuối (End-stage renal disease - ESRD).
Các triệu chứng bệnh thận gồm:
- Màu nước tiểu bất thường, tiểu ra máu
- Đau mạn sườn
- Huyết áp cao
- Phù toàn thân
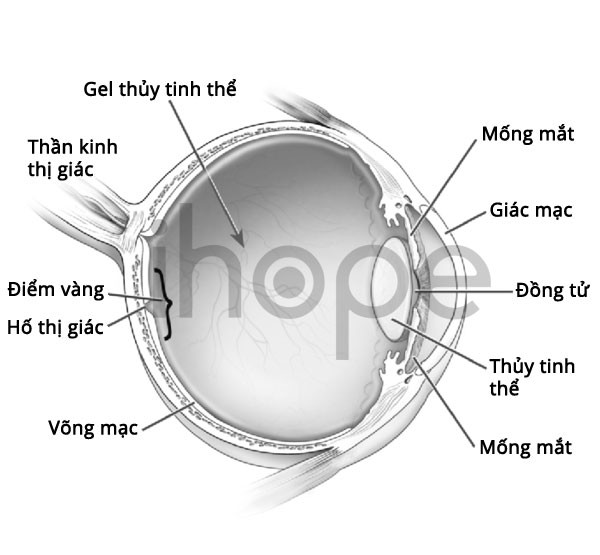
Suy giảm thính lực cũng là triệu chứng đặc trưng của hội chứng Alport do bất thường trong ống tai, biểu hiện ở cuối thời kỳ sơ sinh hoặc đầu tuổi vị thành niên. Người bệnh cũng có thể gặp các vấn đề về mắt như suy giảm thị lực, đục thuỷ tinh thể, rối loạn cảm nhận màu sắc ở võng mạc, trường hợp nghiêm trọng hiếm gặp dẫn tới mù lòa.
Nguồn: U.S. National Library of Medicine
Độ phổ biến
Người ta ước tính tỷ lệ mắc hội chứng Alport khoảng 1/50.000 trẻ sơ sinh.
Nguyên nhân
Hội chứng Alport do đột biến gen COL4A3, COL4A4 và COL4A5 gây ra. Những gen này cung cấp hướng dẫn tạo ra một thành phần của protein gọi là collagen loại IV. Chúng tham gia vào cấu trúc của cầu thận. Cầu thận là những cụm mạch máu chuyên biệt có chức năng loại bỏ nước, chất cặn bã ra khỏi máu và tạo ra nước tiểu. Đột biến gen gây ra bất thường trong quá trình lọc và loại bỏ nước, chất cặn ra khỏi máu, đồng thời để máu và protein đi vào nước tiểu. Điều này dẫn đến suy thận - triệu chứng đặc trưng của hội chứng Alport.
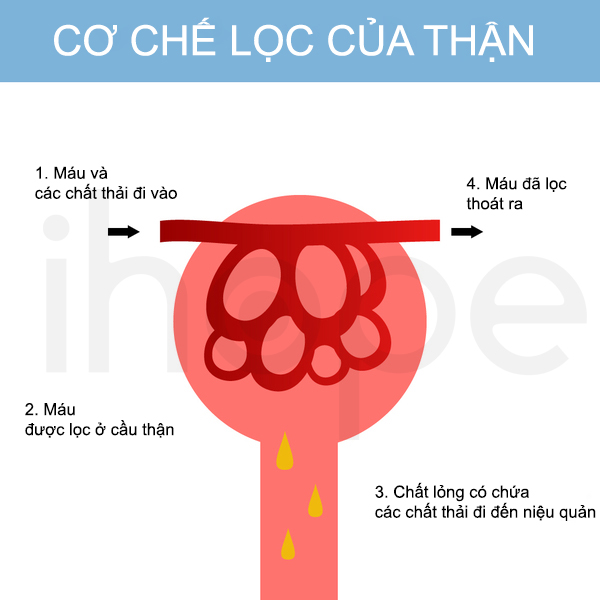
Nguồn: Mr. High Sky/Shutterstock.com
Collagen loại IV cũng là thành phần quan trọng của cấu trúc tai trong, đặc biệt là cơ quan Corti, biến đổi sóng âm thành thành xung thần kinh cho não. Khi đột biến các gen liên quan đến hội chứng Alport dẫn tới tạo collagen loại IV bất thường làm người bệnh mất thính lực.
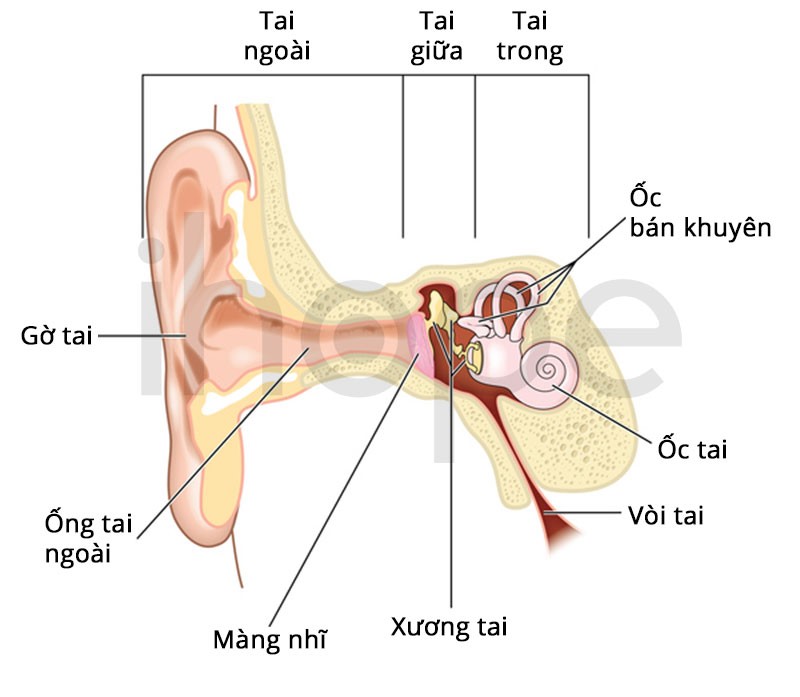
Nguồn: Shutterstock
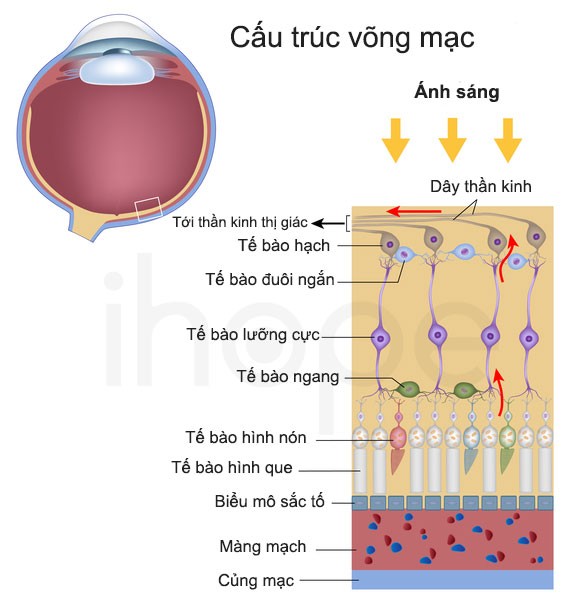
Trong mắt, collagen loại IV có chức năng duy trì hình dạng của thuỷ tinh thể và màu sắc của võng mạc. Các đột biến làm phá vỡ collagen loại IV có thể dẫn đến thủy tinh thể bị giãn và võng mạc có màu bất thường.
Nguồn: National Institutes of Health
Chẩn đoán
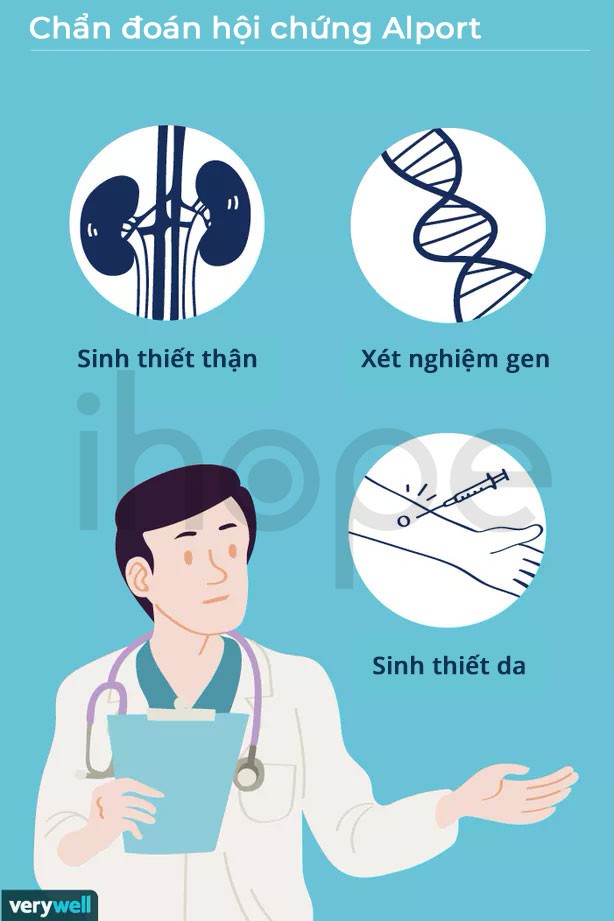
Chẩn đoán hội chứng Alport dựa trên tiền sử mắc bệnh của gia đình, triệu chứng và kết quả xét nghiệm lâm sàng. Người bị nghi ngờ mắc bệnh cần kiểm tra thêm thị lực và thính giác. Có 2 loại xét nghiệm chẩn đoán, bao gồm:
- Sinh thiết thận hoặc da. Nếu người bệnh bị suy giảm chức năng thận không rõ nguyên nhân, tiểu ra máu, protein niệu cần làm sinh thiết thận. Mục đích nhằm phát hiện dấu hiệu thận yếu (dựa vào lượng máu), bệnh gây ảnh hưởng đến thận như tiểu đường.
- Xét nghiệm di truyền. Xét nghiệm dùng để phát hiện đột biến gen có thể dẫn đến hội chứng Alport.
Chẩn đoán sớm hội chứng Alport là bắt buộc, bởi vì các biến chứng của bệnh thường biểu hiện ở thời thơ ấu hoặc đầu tuổi trưởng thành. Nếu không bắt đầu điều trị kịp thời, bệnh thận có thể diễn biến nghiêm trọng ở tuổi trưởng thành.
Các loại xét nghiệm khác có thể dùng để loại trừ các bệnh khác trong danh sách chẩn đoán, đánh giá tình trạng của bệnh nhân hoặc nghi ngờ ban đầu hội chứng Alport. Tuy nhiên, đây không phải là xét nghiệm chẩn đoán, bao gồm:
- Xét nghiệm nước tiểu. Kiểm tra mẫu nước tiểu bằng kính hiển vi nhằm phát hiện thành phần bất thường. Nếu phát hiện protein, nitrit, huyết sắc tố thì có thể chẩn đoán mắc bệnh sỏi thận, nhiễm trùng đường tiết niệu, tiểu đường hoặc ung thư thận.
- Xét nghiệm máu toàn phần. Đo nồng độ hemoglobin hoặc hematocrit trong máu chẩn đoán bệnh thận mãn tính.
- Xét nghiệm creatinine máu. đo mức độ creatinine trong máu kiểm tra tình trạng thận. Nồng độ creatinine cao hơn mức bình thường là dấu hiệu tắc nghẽn đường tiết niệu, suy thận hoặc nhiễm trùng máu.
- Xét nghiệm nitơ urê máu (Blood urea nitrogen - BUN). Đo lượng nitơ, ure trong máu để kiểm tra chức năng thận. Nếu bệnh nhân có lượng chất này cao hơn bình thường thì có thể chẩn đoán mắc bệnh viêm cầu thận, suy thận, nhiễm trùng thận hoặc hoại tử ống thận cấp tính.
Nguồn: Verywell Health
Điều trị
Hiện nay chưa có phương pháp điều trị hội chứng Alport mà chỉ tập trung điều trị triệu chứng cụ thể ở mỗi người bệnh và làm chậm tiến triển của bệnh thận.
Bệnh thận
Một số loại thuốc như:
- Thuốc ức chế men chuyển (ACE) hoặc thuốc chẹn thụ thể angiotensin để giảm huyết áp của bạn và có khả năng làm giảm protein trong nước tiểu và làm chậm sự tiến triển của bệnh thận
- Chế độ ăn hạn chế muối
- Thuốc nước còn được gọi là thuốc lợi tiểu
- Chế độ ăn ít protein
Tuy nhiên, nhiều bệnh nhân tiến triển thành bệnh thận giai đoạn cuối, do đó họ sẽ phải chạy thận hoặc ghép thận.
- Chạy thận là một quá trình nhân tạo để loại bỏ và lọc chất thải ra khỏi cơ thể bằng máy. Máy lọc máu về cơ bản có chức năng thay thế thận.
- Ghép thận bao gồm phẫu thuật thay thế quả thận bị hỏng bằng một quả thận khỏe mạnh từ người hiến tặng.
Cao huyết áp
Bác sĩ sẽ kê đơn thuốc để giúp kiểm soát huyết áp, bao gồm thuốc ức chế men chuyển, thuốc chẹn beta và thuốc chẹn kênh canxi. Những chất này giúp giảm nguy cơ phát triển bệnh tim và cũng làm chậm sự tiến triển của bệnh thận.
Bệnh mắt
Nếu người bệnh có hình dạng thủy tinh thể bất thường, bác sĩ có thể cho đeo kính hoặc phẫu thuật đục thủy tinh thể. Các đốm trắng trong mắt không ảnh hưởng đến thị lực, vì vậy thông thường không cần điều trị.
Mất thính giác
Nếu bị mất thính giác do hội chứng Alport, người bệnh rất có thể bị điếc vĩnh viễn. Tuy nhiên, máy trợ thính là một biện pháp hỗ trợ hiệu quả cho người bệnh.
Dạng di truyền
Hội chứng Alport có nhiều kiểu di truyền khác nhau. Khoảng 80% trường hợp do đột biến gen COL4A5, di truyền theo kiểu liên kết với nhiễm sắc thể giới tính X. Nam giới chỉ có một nhiễm sắc thể X, do đó một bản sao của gen COL4A5 đột biến trong mỗi tế bào đủ gây ra suy thận và các triệu chứng nghiêm trọng khác của bệnh. Người cha bị bệnh không truyền tính trạng này cho con trai của họ. Ở phụ nữ có hai nhiễm sắc thể X, đột biến ở một bản sao của gen COL4A5 thường chỉ gây ra chứng tiểu máu, tuy nhiên một số phụ nữ bị bệnh có triệu chứng nghiêm trọng hơn.
Khoảng 15% trường hợp mắc hội chứng Alport do đột biến ở cả hai bản sao của gen COL4A3 hoặc COL4A4 và di truyền theo kiểu lặn trên nhiễm sắc thể thường. Một số trường hợp mang gen lặn biểu hiện triệu chứng ít nghiêm trọng hơn như bệnh thận màng đáy mỏng đặc trưng bởi tình trạng tiểu ra máu.

Nguồn: U.S. National Library of Medicine
Khoảng 5% trường hợp mắc bệnh do di truyền trội trên nhiễm sắc thể thường. Những người mắc dạng hội chứng Alport này có một đột biến gen COL4A3 hoặc COL4A4 trong mỗi tế bào. Vẫn chưa rõ lý do phát sinh đột biến ở các gen.

Nguồn: U.S. National Library of Medicine
Phòng ngừa
Hiện nay chưa có phương pháp phòng ngừa hội chứng Alport. Các cặp vợ chồng trước khi mang thai cần tư vấn và xét nghiệm di truyền đảm bảo sinh con khỏe mạnh.
Trường hợp di truyền lặn đột biến gen COL4A3 hoặc COL4A4, cha mẹ mang đột biến dị hợp nên gần như không có biểu hiện bệnh, do đó rất khó phát hiện cho đến khi sinh con. Để chủ động phòng ngừa, cha mẹ nên làm xét nghiệm sàng lọc gen lặn để chủ động cho tương lai của con.
Đồng thời, nhận thức yếu tố nguy cơ như tiền sử bệnh trong gia đình cho phép tầm soát bệnh tốt hơn.
Các tên gọi khác
- Congenital hereditary hematuria
- Hematuria-nephropathy-deafness syndrome
- Hematuric hereditary nephritis
- Hemorrhagic familial nephritis
- Hemorrhagic hereditary nephritis
- Hereditary familial congenital hemorrhagic nephritis
- Hereditary hematuria syndrome
- Hereditary nephritis
- hereditary nephritis with sensory deafness
References
- Genetic Testing Information. Alport syndrome. Retrieved June 15, 2022 from https://www.ncbi.nlm.nih.gov/gtr/conditions/C1567741/
- Genetic Testing Information. X-linked Alport syndrome. Retrieved June 15, 2022 from https://www.ncbi.nlm.nih.gov/gtr/conditions/C4746986/
- Genetic Testing Information. Autosomal dominant Alport syndrome. Retrieved June 15, 2022 from https://www.ncbi.nlm.nih.gov/gtr/conditions/C4746547/
- Genetic Testing Information. Autosomal recessive Alport syndrome. Retrieved June 15, 2022 from https://www.ncbi.nlm.nih.gov/gtr/conditions/C4746745/
- Catalog of Genes and Diseases from OMIM. ALPORT SYNDROME 1, X-LINKED; ATS1. Retrieved June 15, 2022 from https://omim.org/entry/301050
- Catalog of Genes and Diseases from OMIM. ALPORT SYNDROME 2, AUTOSOMAL RECESSIVE; ATS2. Retrieved June 15, 2022 from https://omim.org/entry/203780
- Catalog of Genes and Diseases from OMIM. ALPORT SYNDROME 3, AUTOSOMAL DOMINANT; ATS3. Retrieved June 15, 2022 from https://omim.org/entry/104200
- Genetic and Rare Diseases Information Center. Alport syndrome. Retrieved June 15, 2022 from https://rarediseases.info.nih.gov/diseases/5785/alport-syndrome
- U.S National Library of Medicine. Alport syndrome. Retrieved June 15, 2022 from https://medlineplus.gov/genetics/condition/alport-syndrome/
- Genetic and Rare Diseases Information Center . Retrieved June 15, 2022 from https://rarediseases.org/rare-diseases/alport-syndrome/
- Verywell health. An Overview Of Alport Syndrome. Retrieved June 15, 2022 from https://www.verywellhealth.com/alport-syndrome-overview-4176361
- National Kidney Foundation. Alport Syndrome. Retrieved June 15, 2022 from https://www.kidney.org/atoz/content/alport





