
Bệnh Charcot-Marie-Tooth (Charcot-Marie-Tooth Disease – CMT) là bệnh di truyền xảy ra do tổn thương các dây thần kinh ngoại vi kết nối não và tủy sống với các cơ và tế bào cảm giác phát hiện ra các cảm giác như chạm, đau, nóng và âm thanh. Những tổn thương này tiến triển nặng hơn theo thời gian, người bệnh có thể bị thay đổi hoặc mất cảm giác và teo các cơ ở bàn chân, cẳng chân và bàn tay.
Biểu hiện lâm sàng
Bệnh Charcot-Marie-Tooth có thể khởi phát từ khi còn nhỏ và biểu hiện rõ ràng tại tuổi thiếu niên. Các triệu chứng của bệnh khác nhau tùy theo mức độ nghiêm trọng và độ tuổi khởi phát ngay cả giữa các thành viên trong cùng một gia đình. Một số người không bao giờ nhận ra mình mắc bệnh vì các triệu chứng của họ rất nhẹ và hầu hết khiếm khuyết thể chất với mức độ vừa phải. Một tỷ lệ nhỏ người bệnh bị suy nhược nghiêm trọng hoặc có thể đe dọa đến tính mạng. Tuy nhiên, đa số người mắc bệnh Charcot-Marie-Tooth có tuổi thọ bình thường.
Thông thường, các triệu chứng sớm của bệnh Charcot-Marie-Tooth do teo cơ bàn chân. Người bệnh có các bất thường chân như vòm cao, bàn chân bẹt hoặc ngón chân cong. Họ gặp khó khăn khi gập bàn chân hoặc đi bằng gót chân, nên bước dài hơn bình thường. Do đó, họ tăng nguy cơ chấn thương mắt cá chân và vấp ngã. Theo thời gian, các cơ cẳng chân yếu dần nhưng hiếm khi người bệnh phải sử dụng xe lăn.
Những người mắc bệnh cũng có thể bị yếu tay. Triệu chứng này gây khó khăn cho các hoạt động hàng ngày như viết, thắt nút và xoay nắm cửa. Họ thường giảm nhạy cảm với xúc giác, nóng lạnh tại bàn chân và cẳng chân nhưng đôi khi cảm thấy đau nhức hoặc bỏng rát. Trong một số ít trường hợp, người bệnh mất thị lực hoặc mất thính lực.
Độ phổ biến
Bệnh Charcot-Marie-Tooth là bệnh di truyền phổ biến liên quan đến các dây thần kinh ngoại biên. Người ta ước tính tỉ lệ mắc bệnh khoảng 1/3.300 người.
Nguyên nhân
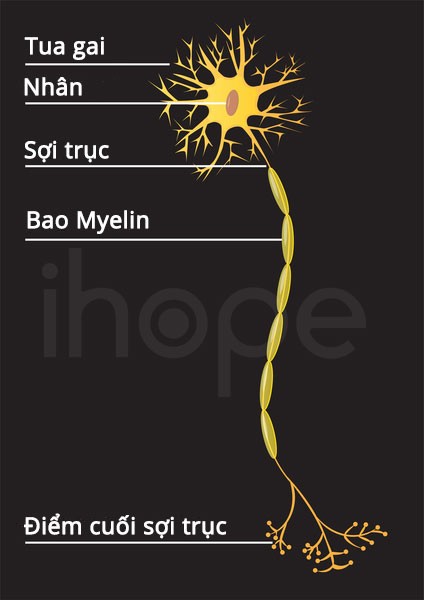
Bệnh Charcot-Marie-Tooth do đột biến tại nhiều gen khác nhau. Những gen này cung cấp hướng dẫn tạo ra các protein liên quan đến chức năng của dây thần kinh ngoại vi ở bàn chân, cẳng chân và bàn tay. Cơ chế ảnh hưởng của các đột biến gen liên quan đến chức năng của protein chưa được tìm hiểu rõ. Tuy nhiên, chúng có thể làm suy giảm các sợi trục, nơi truyền các xung thần kinh hoặc ảnh hưởng đến các tế bào chuyên biệt sản xuất myelin. Trong hầu hết các trường hợp, các dây thần kinh dài truyền xung động đến các phần phụ của cơ thể có nhiều khả năng bị ảnh hưởng hơn. Do đó, các tế bào thần kinh ngoại vi từ từ mất khả năng kích thích các cơ ở bàn chân, cẳng chân, bàn tay và truyền tín hiệu cảm giác từ các phần phụ này đến não. Các đột biến khác nhau trong cùng một gen có thể gây ra những dấu hiệu và triệu chứng với mức độ nghiêm trọng khác nhau hoặc dẫn đến các dạng bệnh Charcot-Marie-Tooth khác nhau.
Nguồn: National Library of Medicine
Bệnh Charcot-Marie-Tooth loại 1
Nguyên nhân do bất thường trong vỏ myelin, bao gồm sáu dạng phụ:
- Loại 1A do lặp đoạn trên nhiễm sắc thể số 17 tạo thêm một bản sao gen PMP22 trong mỗi tế bào. Protein PMP22 là thành phần quan trọng của vỏ myelin. Gen PMP22 biểu hiện quá mức gây bất thường cấu trúc và chức năng của vỏ myelin.
- Loại 1B do đột biến gen mang chỉ thị sản xuất protein MPZ, còn gọi là P0 - thành phần quan trọng khác của vỏ myelin. Hầu hết các đột biến thuộc loại đột biến điểm. Đến nay, người ta đã xác định được hơn 120 đột biến điểm khác nhau trong gen P0.
- Các nguyên nhân khác ít phổ biến hơn gây bệnh do đột biến trong gen SIMPLE (còn gọi là LITAF), EGR2 và NEFL.
Bệnh Charcot-Marie-Tooth loại 2
Nguyên nhân do đột biến gen MFN2 dẫn đến bất thường trong sợi trục của tế bào thần kinh ngoại vi, ít gặp hơn loại 1. Một số bệnh nhân loại 2 có thể ảnh hưởng đến dây thanh âm hoặc dây thần kinh phrenic gây ra các vấn đề về giọng nói hoặc hô hấp.
Bệnh Charcot-Marie-Tooth loại 3
Còn gọi là bệnh Dejerine-Sottas, bắt đầu từ giai đoạn sơ sinh. Trẻ mắc bệnh bị teo cơ nghiêm trọng, yếu ớt, chậm phát triển kỹ năng vận động và các vấn đề về cảm giác. Các triệu chứng có thể tiến triển thành tàn tật nghiêm trọng, mất cảm giác và cong cột sống. Rối loạn hiếm gặp này do đột biến tại nhiều gen, bao gồm PMP22, MPZ, EGR2 và di truyền theo kiểu trội hoặc lặn.
Bệnh Charcot-Marie-Tooth loại X
Nguyên nhân do đột biến gen GJB1. Gen này cung cấp hướng dẫn tạo ra một loại protein trong tế bào Schwann tạo myelin. Nam giới mang gen đột biến có các triệu chứng nặng hơn nữ giới.
Một số tên gọi khác khác được sử dụng để chỉ các dạng cụ thể của bệnh Charcot-Marie-Tooth. Ví dụ, hội chứng Roussy-Levy là một dạng của CMT11, người bệnh thường có thêm biểu hiện run. Hội chứng Dejerine-Sottas là dạng bệnh Charcot-Marie-Tooth nặng (đôi khi gọi là loại 3 - CMT3) ở trẻ dưới 8 tuổi. CMTX5 còn gọi là hội chứng Rosenberg-Chutorian.
Danh sách các gen liên quan đến bệnh Charcot Marie Tooth vẫn đang được nghiên cứu và làm rõ.
Chẩn đoán
Người bệnh được kiểm tra sức khỏe, thăm hỏi tiền sử bệnh và tìm các dấu hiệu lâm sàng như:
- Khó nhấc bàn chân lên và thực hiện chuyển động bằng ngón chân
- Thiếu phản xạ co duỗi ở chân
- Mất kiểm soát và teo cơ bàn chân hoặc cẳng chân
- Các bó dây thần kinh dưới da chân dày
Các xét nghiệm kiểm tra dẫn truyền thần kinh, đo điện cơ.
Sinh thiết dây thần kinh. Hình dạng dây thần kinh của người mắc bệnh có thể khác với các dây thần kinh bình thường khi quan sát dưới kính hiển vi.
Xét nghiệm di truyền có thể phát hiện bệnh Charcot-Marie-Tooth, tuy nhiên không thể phát hiện tất cả các loại.
Điều trị
Hiện nay, chưa có phương pháp điều trị bệnh Charcot-Marie-Tooth. Một số phương pháp hỗ trợ giúp người bệnh cải thiện cuộc sống như:
- Phẫu thuật chỉnh hình hoặc thiết bị hỗ trợ (ví dụ như nẹp hoặc giày chỉnh hình) điều trị các biến dạng khớp và bàn chân nghiêm trọng giúp bệnh nhân đi lại dễ dàng hơn.
- Liệu pháp vật lí và vận động giúp duy trì sức mạnh cơ bắp và cải thiện chức năng hoạt động độc lập.
Dạng di truyền
Kiểu di truyền thay đổi theo loại bệnh Charcot Marie Tooth:
- Bệnh Charcot-Marie-Tooth loại 1, hầu hết các trường hợp loại 2 và các dạng trung gian di truyền theo kiểu trội trên nhiễm sắc thể thường. Do đó, một bản sao của gen đột biến trong mỗi tế bào đủ gây ra bệnh. Nếu bố hoặc mẹ mắc bệnh, nguy cơ con của họ mắc bệnh là 50%.
- Bệnh Charcot-Marie-Tooth loại 4, một số kiểu phụ của loại 2 và một số dạng trung gian di truyền theo kiểu lặn trên nhiễm sắc thể thường. Do đó, cả hai bản sao của gen đột biến trong mỗi tế bào mới có khả năng gây bệnh. Thông thường, bố mẹ của người bệnh mang bản sao của gen đột biến nhưng không biểu hiện dấu hiệu và triệu chứng của bệnh.
- Bệnh Charcot-Marie-Tooth loại X di truyền theo kiểu trội liên kết nhiễm sắc thể giới tính X. Trong hầu hết các trường hợp, những người đàn ông mang gen bệnh đều mắc bệnh và biểu hiện triệu chứng nghiêm trọng hơn phụ nữ mắc bệnh. Do di truyền liên kết X nên người cha không thể truyền tính trạng liên kết X cho con trai của họ. Tất cả con gái của những người đàn ông bị bệnh sẽ có một nhiễm sắc thể X mang gen đột biến nhưng họ có thể chỉ có các triệu chứng nhẹ.

Nguồn: U.S. National Library of Medicine

Nguồn: U.S. National Library of Medicine

Nguồn: U.S. National Library of Medicine
Một số trường hợp mắc bệnh Charcot-Marie-Tooth là kết quả của đột biến mới và xảy ra ở người không có tiền sử mắc bệnh trong gia đình họ.
Phòng ngừa
Với các dạng bệnh Charcot-Marie-Tooth di truyền lặn trên nhiễm sắc thể thường, cha mẹ mang đột biến dị hợp nên gần như không có biểu hiện bệnh, do đó rất khó phát hiện cho đến khi sinh con. Để chủ động phòng ngừa, cha mẹ nên làm xét nghiệm sàng lọc gen lặn để chủ động cho tương lai của con. Các thành viên trong gia đình nên khám sức khỏe và tầm soát bệnh định kỳ nếu có thành viên mắc bệnh. Các cặp vợ chồng trước khi mang thai cần tư vấn và xét nghiệm di truyền đảm bảo sinh con khỏe mạnh.
Các tên gọi khác
- Charcot-Marie-Tooth hereditary neuropathy
- Charcot-Marie-Tooth syndrome
- CMT
- Hereditary motor and sensory neuropathy
- HMSN
- Peroneal muscular atrophy
- PMA
- Charcot-Marie-Tooth Disease
References
- Genetic Testing Information. Charcot-Marie-Tooth disease. Retrieved August 16, 2022 from https://www.ncbi.nlm.nih.gov/gtr/conditions/C0007959/
- Genetic and Rare Diseases Information Center. Charcot-Marie-Tooth disease. Retrieved Aug 16, 2022 from https://rarediseases.info.nih.gov/diseases/6034/charcot-marie-tooth-disease
- Catalog of Genes and Diseases from OMIM. CHARCOT-MARIE-TOOTH DISEASE AND DEAFNESS. Retrieved Aug 16, 2022 from https://omim.org/entry/118300
- U.S National Library of Medicine. Charcot-Marie-Tooth disease. Retrieved Aug 16, 2022 from https://medlineplus.gov/genetics/condition/charcot-marie-tooth-disease/
- National Human Genome Research Institute. Charcot-Marie-Tooth disease. Retrieved Aug 22, 2022 from https://www.genome.gov/Genetic-Disorders/Charcot-Marie-Tooth-Disease
- U.S National Library of Medicine. Charcot-Marie-Tooth disease. Retrieved Aug 22, 2022 from https://medlineplus.gov/ency/article/000727.htm
- National Institute of Neurological Disorders and Stroke. Charcot-Marie-Tooth Disease Fact Sheet. Retrieved Aug 22, 2022 from https://www.ninds.nih.gov/charcot-marie-tooth-disease-fact-sheet
- Johns Hopkins Medicine. Charcot-Marie-Tooth disease. Retrieved Aug 22, 2022 from https://www.hopkinsmedicine.org/health/conditions-and-diseases/charcotmarietooth-disease
- National Health Service. Charcot-Marie-Tooth disease. Retrieved Aug 22, 2022 from https://www.nhs.uk/conditions/charcot-marie-tooth-disease/
- Mayo Foundation for Medical Education and Research. Charcot-Marie-Tooth disease. Retrieved Aug 22, 2022 from https://www.mayoclinic.org/diseases-conditions/charcot-marie-tooth-disease/symptoms-causes/syc-20350517
- Muscular Dystrophy Association. Charcot-Marie-Tooth disease. Retrieved Aug 22, 2022 from https://medlineplus.gov/charcotmarietoothdisease.html





