Hội chứng mất đoạn 1p21.3 (1p21.3 microdeletion syndrome) là một bất thường di truyền hiếm gặp bắt nguồn từ thay đổi cấu trúc nhiễm sắc thể. Người bệnh có biểu hiện gồm thiểu năng trí tuệ, hành vi bất thường và khuôn mặt dị biệt.
Biểu hiện lâm sàng
Trí tuệ và hành vi
Người mắc hội chứng mất đoạn 1p21.3 thường gặp các vấn đề về trí tuệ và hành vi như:
- Chậm phát triển khả năng nói và ngôn ngữ
- Thiểu năng trí tuệ
- Rối loạn phổ tự kỉ
- Ăn nhiều bất thường
- Hung hăng
- Có hành vi tự làm hại bản thân
Khuôn mặt dị biệt
Người bệnh có khuôn mặt dị biệt với các biểu hiện đặc trưng bao gồm:
- Đầu to
- Tai dài
- Mắt trũng sâu
- Mũi ngắn, đầu mũi lớn
- Môi dưới dày
- Má đầy đặn
- Hàm nhỏ, miệng rộng
- Mắt xếch
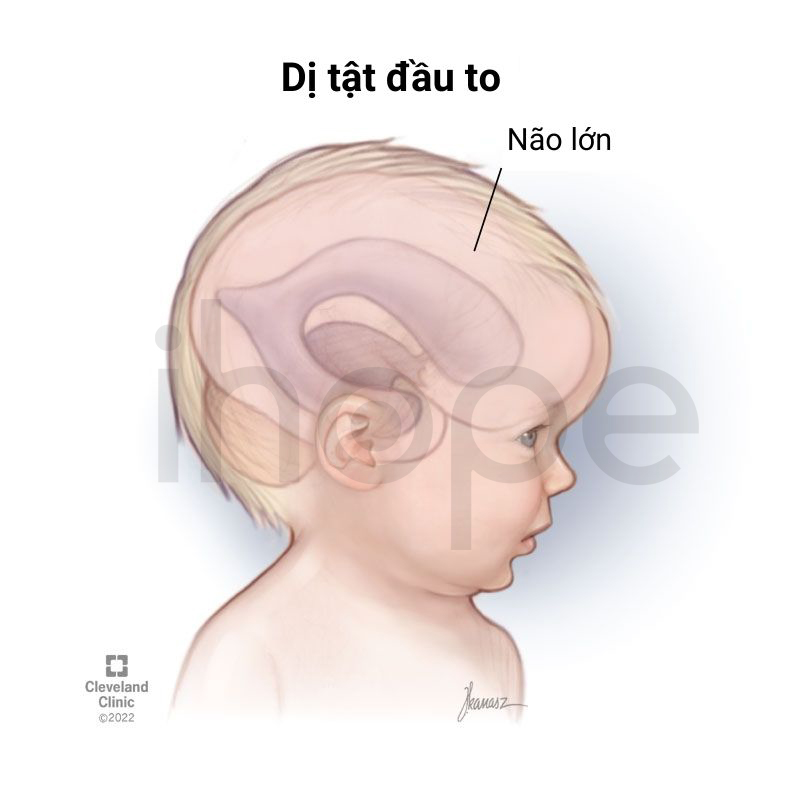
Ảnh: Dị tật đầu to
Nguồn: Cleveland Clinic
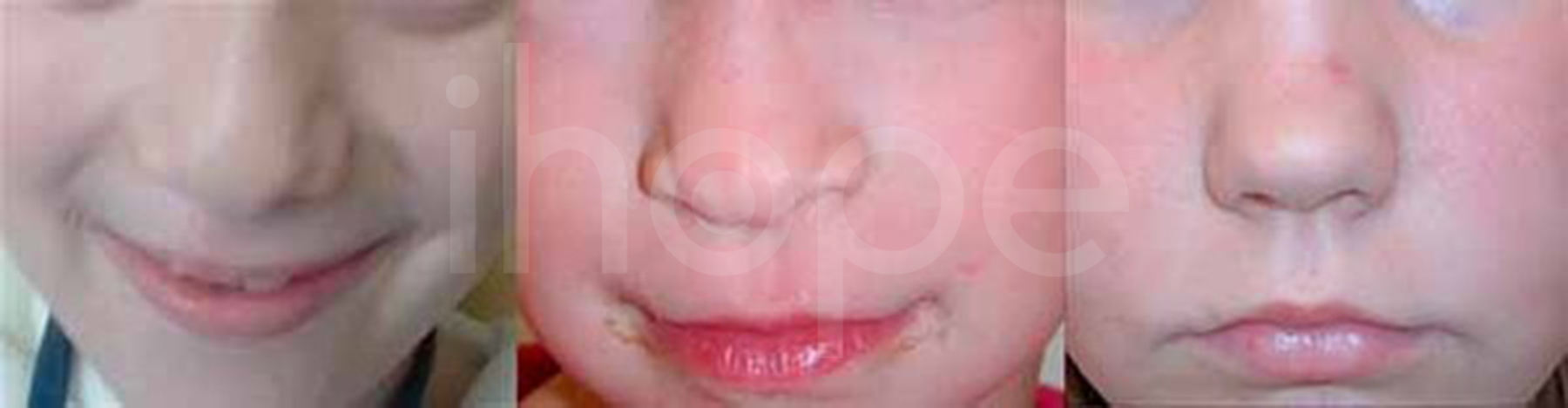
Ảnh: Mũi ngắn với đầu mũi lớn
Nguồn: Elements of Morphology, National Human Genome Research Institute
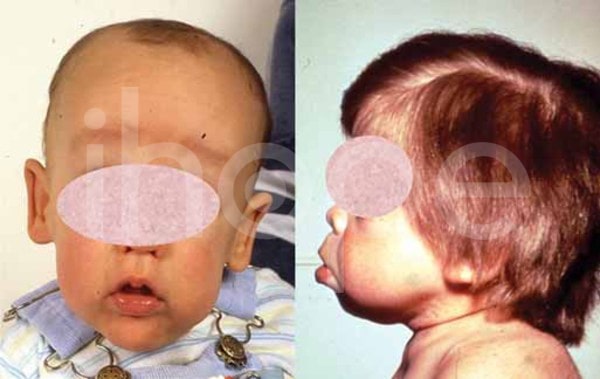
Ảnh: Má đầy đặn
Nguồn: Elements of Morphology, National Human Genome Research Institute
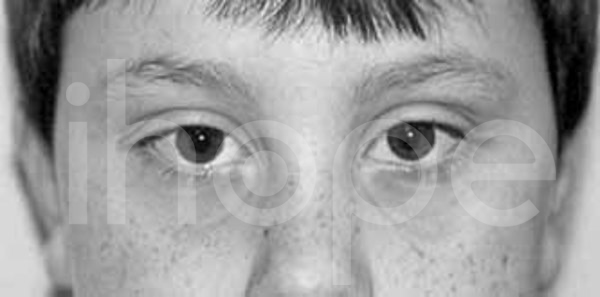
Ảnh: Khe mi mắt xếch ngược lên
Nguồn: National Human Genome Research Institute
Các triệu chứng khác
Hội chứng mất đoạn 1p21.3 có thể ảnh hưởng đến thị lực của người bệnh, dẫn đến nhiều tật khúc xạ như loạn thị và cận thị. Trong một số trường hợp, bệnh nhân có thể mắc chứng khớp dẻo khiến các khớp di chuyển linh hoạt bất thường.
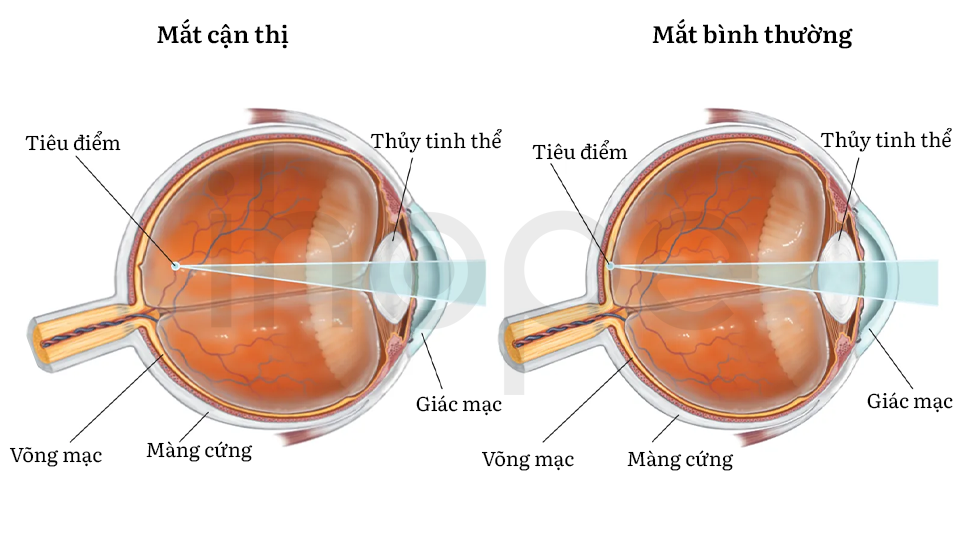
Nguồn: Encyclopaedia Britannica
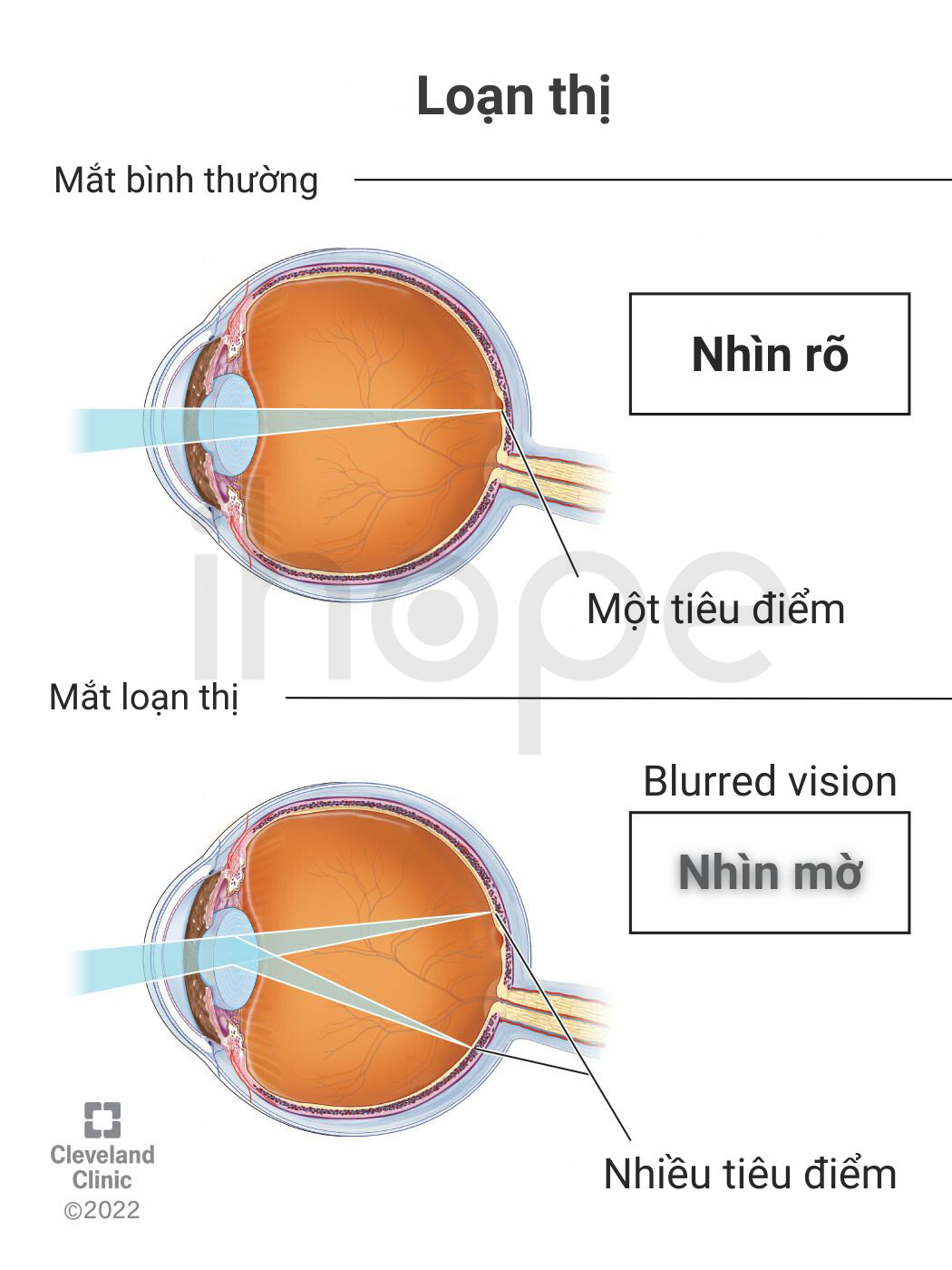
Ảnh: Loạn thị
Nguồn: Cleveland Clinic
Độ phổ biến
Hội chứng mất đoạn 1p21.3 rất hiếm gặp, ước tính tỉ lệ mắc bệnh trên toàn thế giới chưa đến 1/1.000.000 người. Hiện nay, người ta ghi nhận chưa đến 10 trường hợp mắc bệnh.
Nguyên nhân
Đột biến mất đoạn nhỏ thuộc vùng p21.3 trên nhánh ngắn của nhiễm sắc thể 1 gây ra hội chứng mất đoạn 1p21.3. Đột biến làm mất một bản sao của gen DYPD và MIR137.
Gen DYPD cung cấp hướng dẫn tạo ra enzyme dihydropyrimidine dehydrogenase có chức năng huỷ uracil và thymine không cần thiết. Uracil và thymine là hai đơn vị cấu tạo nên ARN. Đối với người mắc hội chứng mất đoạn 1p21.3, enzyme dihydropyrimidine dehydrogenase giảm hoạt động dẫn đến lượng uracil và thymine dư thừa trong máu, nước tiểu, dịch não tuỷ. Uracil và thymine tích tụ trong dịch não tủy có thể gây ra các triệu chứng như động kinh, chậm phát triển tâm thần, hoặc các bệnh lí thần kinh khác. Người ta đã ghi nhận các trường hợp bệnh nhân bị thiểu năng trí tuệ do mất một phần hoặc toàn bộ gen DYPD.
Nguồn: U.S. National Library of Medicine
Gen MIR137 cung cấp hướng dẫn tạo ra miR-137 có chức năng điều hoà biểu hiện gen. miR-137 là đoạn ARN ngắn và không mã hóa protein. Chúng tham gia điều hòa biểu hiện gen bằng cách liên kết với vùng không dịch mã của ARN thông tin mục tiêu, sau đó ngăn cản quá trình dịch mã hoặc làm phân hủy ARN thông tin này.
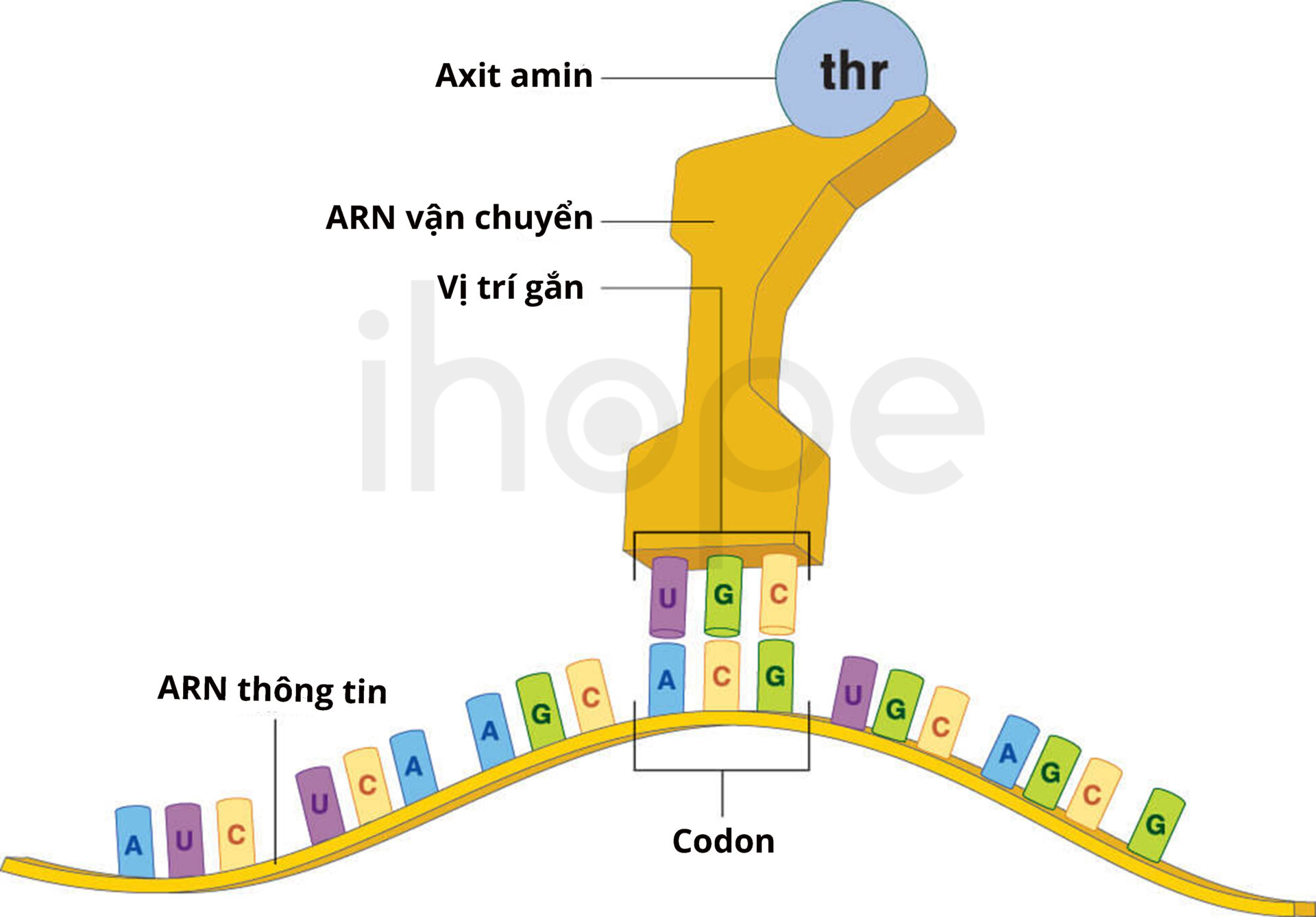
Nguồn: Biology Dictionary
Gen MIR137 hiện diện chủ yếu tại các khớp thần kinh synap trong vỏ não và vùng hồi hải mã, chúng gia tăng biểu hiện trong quá trình phát triển não và biệt hoá tế bào thần kinh. Vì vậy, đột biến mất gen MIR137 có thể ảnh hưởng đến khả năng hoạt động của não bộ.
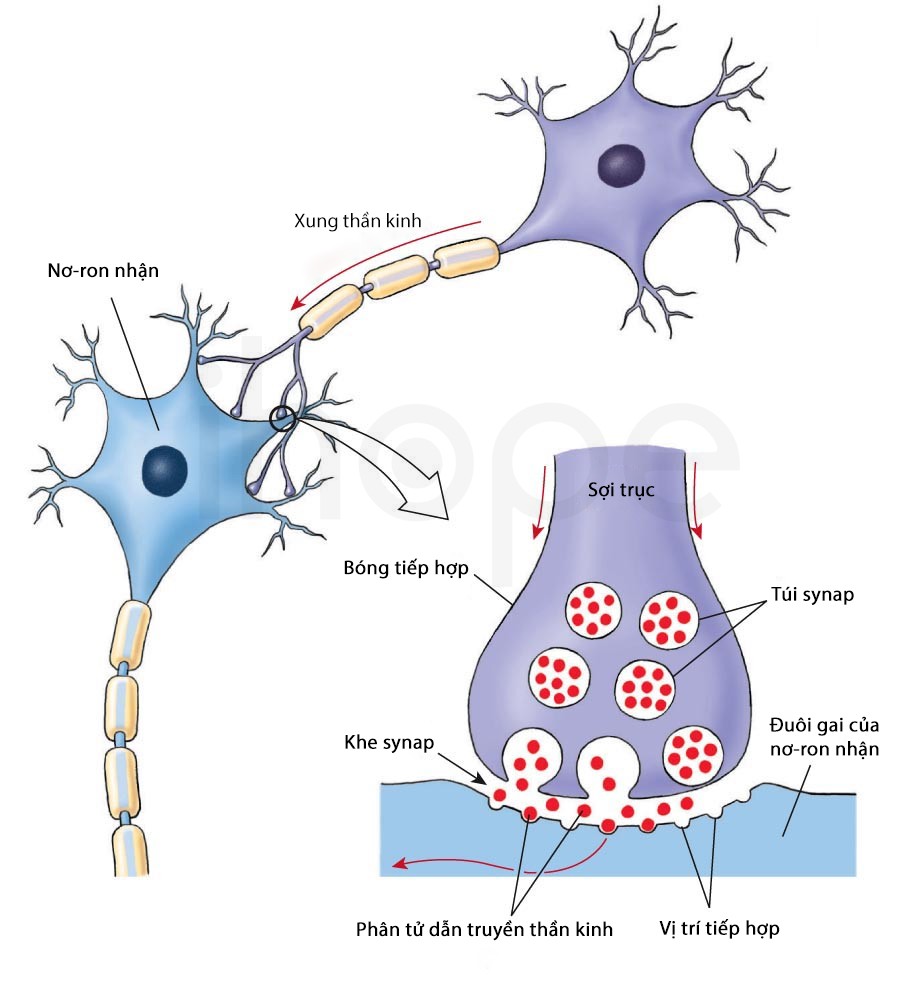
Nguồn: socratic.org
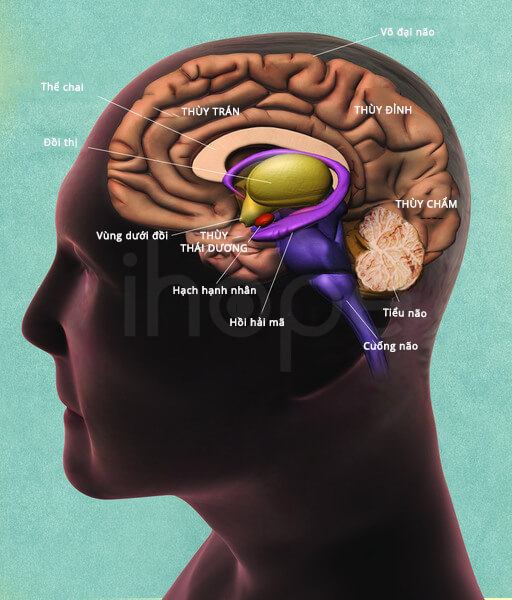
Ảnh: Mặt cắt bên trong của não
Nguồn: National Institutes of Health[/caption]
Chẩn đoán
Hội chứng mất đoạn 1p21.3 được chẩn đoán dựa trên biểu hiện lâm sàng và xét nghiệm di truyền. Phương pháp phân tích nhiễm sắc thể thông thường khó phát hiện các đột biến mất đoạn nhỏ, vì vậy, bệnh nhân có thể thực hiện lai huỳnh quang tại chỗ (Fluorescence In Situ Hybridization - FISH) hoặc lai so sánh bộ gen (array Comparative Genomic Hybridization - aCGH) nhằm xác định các đột biến mất đoạn nhỏ.
Điều trị
Hiện nay, chưa có phương pháp điều trị hoàn toàn hội chứng mất đoạn 1p21.3. Các liệu pháp chủ yếu nhằm giảm nhẹ biểu hiện và cải thiện chất lượng đời sống người bệnh.
Dạng di truyền
Người ta chưa rõ cơ chế di truyền của hội chứng mất đoạn 1p21.3. Phần lớn các trường hợp mắc bệnh do đột biến mới (de novo) xuất hiện trong quá trình hình thành tế bào sinh sản (trứng, tinh trùng) hoặc quá trình phát triển phôi sớm.
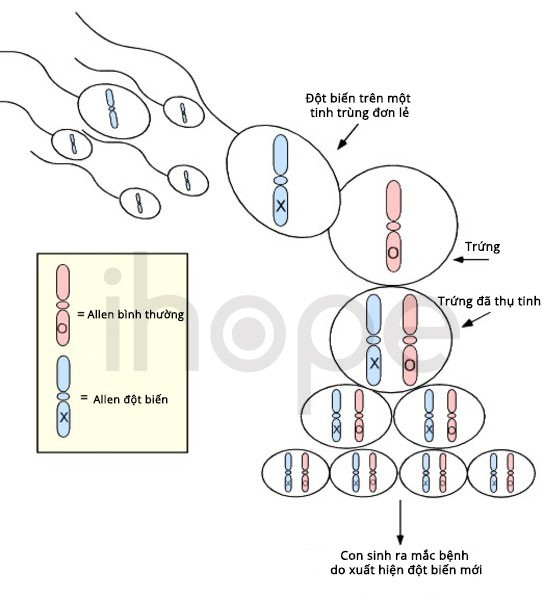
Nguồn: U.S. National Library of Medicine
Phòng ngừa
Hội chứng mất đoạn 1p21.3 do đột biến ngẫu nhiên xảy ra trong quá trình phát triển của thai nhi, vì vậy tất cả thai phụ đều có nguy cơ mang thai bị bệnh. Do đó, sản phụ nên khám thai và siêu âm định kì cũng như làm các xét nghiệm sàng lọc như xét nghiệm NIPT nhằm phát hiện sớm vấn đề có thể xảy ra với thai nhi.
Các tên gọi khác
- Del(1)(p21.3)
- Monosomy 1p21.3
References
- Genetic Testing Information. 1p21.3 microdeletion syndrome. Retrieved September 09, 2024 from https://www.ncbi.nlm.nih.gov/gtr/conditions/C4304578/
- Genetic and Rare Diseases Information Center. 1p21.3 microdeletion syndrome. Retrieved September 09, 2024 from https://rarediseases.info.nih.gov/diseases/21173/1p213-microdeletion-syndrome
- MalaCards. 1p21.3 Microdeletion Syndrome. Retrieved September 09, 2024 from https://www.malacards.org/card/1p213_microdeletion_syndrome
- National Institute of Health. Two New Cases of 1p21.3 Deletions and an Unbalanced Translocation t(8;12) among Individuals with Syndromic Obesity. Retrieved September 09, 2024 from https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4521070/
- National Institute of Health. Association of new deletion/duplication region at chromosome 1p21 with intellectual disability, severe speech deficit and autism spectrum disorder-like behavior: an all-in approach to solving the DPYD enigma. Retrieved September 09, 2024 from https://pubmed.ncbi.nlm.nih.gov/28123791/
- National Organization for Rare Disorders. 1p21.3 microdeletion syndrome. Retrieved September 09, 2024 from https://rarediseases.org/mondo-disease/1p21-3-microdeletion-syndrome/
- Orphanet. 1p21.3 microdeletion syndrome. Retrieved September 09, 2024 from https://www.orpha.net/en/disease/detail/293948