Hội chứng uốn cong động mạch (arterial tortuosity syndrome) ảnh hưởng đến mô liên kết – cấu trúc cung cấp sức mạnh và tính linh hoạt cho cơ thể, bao gồm mạch máu, da, khớp và đường tiêu hóa. Bệnh gây ra hiện tượng mạch máu xoắn và quay bất thường (ngoằn ngoèo). Hiện tượng ngoằn ngoèo phát sinh từ quá trình kéo dài bất thường của các động mạch. Các điểm cuối của động mạch được cố định, phần dài thêm sẽ xoắn và cong. Các bất thường mạch máu khác có thể xảy ra bao gồm hẹp và phình mạch máu, giãn mạch.
Biểu hiện lâm sàng
Người bệnh thường trông già hơn so với tuổi với các đặc điểm khác biệt như khuôn mặt dài , má hóp, khe mắt hẹp với các góc bên ngoài hướng xuống dưới, mũi khoằm bằng sụn mềm, vòm miệng cao , cong, hàm dưới nhỏ và đôi tai lớn. Giác mạc có hình nón và mỏng.
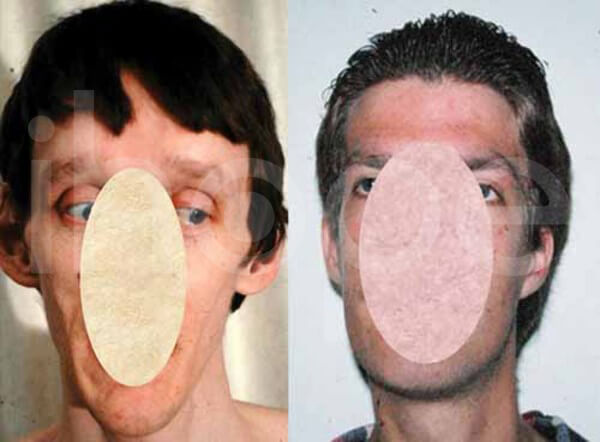
Ảnh: Khuôn mặt dài
Nguồn: National Human Genome Research Institute
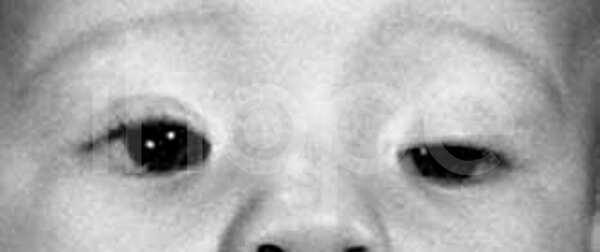
Ảnh: Khe mắt hẹp
Nguồn: National Human Genome Research Institute
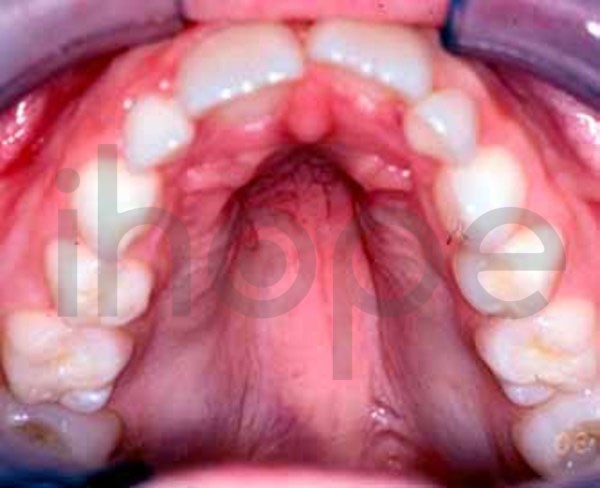
Ảnh: Vòm miệng cao
Nguồn: U.S. National Library of Medicine
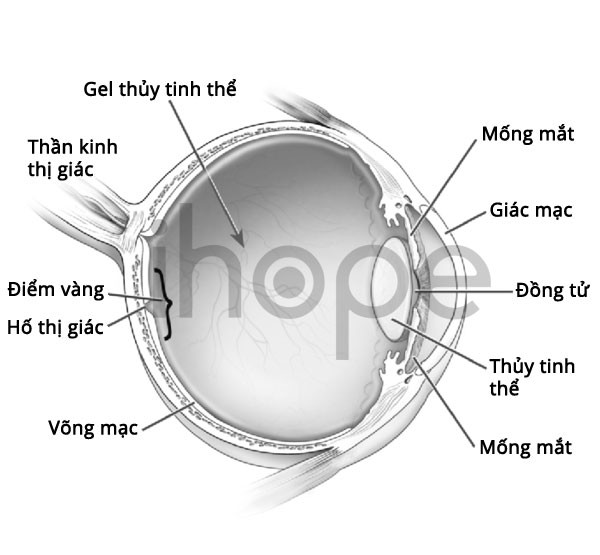
Ảnh: Giải phẫu mắt bình thường
Nguồn: National Institutes of Health
Biến chứng do động mạch bất thường có thể đe dọa tính mạng. Phình hoặc bóc tách đột ngột các lớp trong thành động mạch dẫn đến mất máu ồ ạt từ hệ thống tuần hoàn. Tắc nghẽn làm giảm lưu lượng máu đến các cơ quan quan trọng như tim, phổi hoặc não gây ra các cơn đau tim, các vấn đề về hô hấp và đột quỵ. Hẹp động mạch khiến tim phải làm việc nhiều hơn để bơm máu và có thể dẫn đến suy tim. Do những biến chứng nguy hiểm, hội chứng uốn cong động mạch thường gây tử vong cho trẻ em, tuy nhiên một số trường hợp mắc thể nhẹ có thể sống đến tuổi trưởng thành.
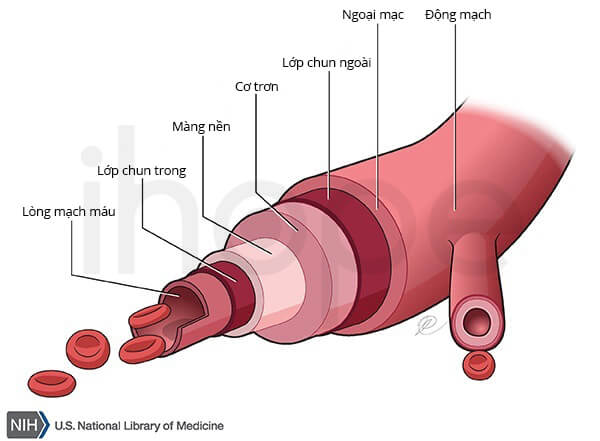
Ảnh: Động mạch
Nguồn: U.S. National Library of Medicine/p>
Đặc điểm của hội chứng uốn cong động mạch ngoài hệ tuần hoàn bao gồm:
- Khớp lỏng lẻo và linh hoạt
- Co rút, hạn chế chuyển động
- Da mềm và co giãn bất thường
- Ngón tay và ngón chân dài, thon
- Vẹo cột sống
- Ngực bị lõm hoặc nhô ra
- Thoát vị qua các túi thừa trong thành ruột
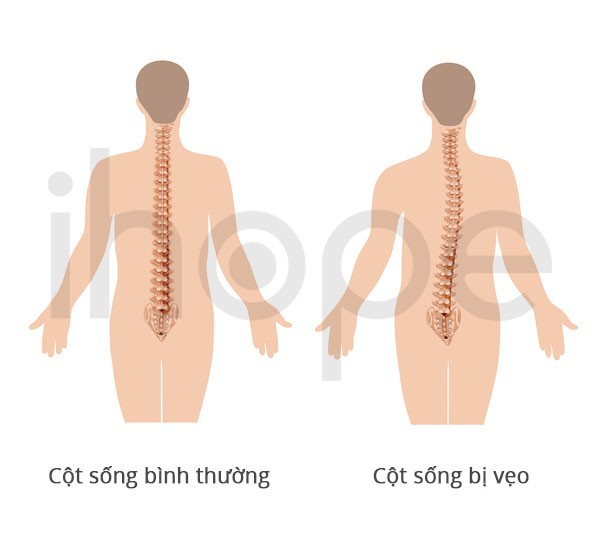
Ảnh: Cột sống bình thường và cột sống bị vẹo.
Nguồn: U.S. National Library of Medicine

Ảnh: Ngực nhô ra
Nguồn: U.S. National Library of Medicine
Độ phổ biến
Hội chứng uốn cong động mạch hiếm gặp, do đó hiện nay tỷ lệ mắc bệnh vẫn chưa được thống kê cụ thể. Khoảng 100 trường hợp mắc bệnh đã báo cáo trong các tài liệu y khoa.
Nguyên nhân
Đột biến gen SLC2A10 gây ra hội chứng uốn cong động mạch. Gen SLC2A10 cung cấp hướng dẫn tạo ra protein GLUT10.Số lượng protein GLUT10 liên quan đến đường tín hiệu yếu tố chuyển đổi tăng trưởng beta (TGF-β). TGF-β tham gia vào quá trình phát triển, phân chia và biệt hóa của tế bào, trong đó có xương, mạch máu, chất nền ngoại bào và xác định cấu trúc, tính chất của các mô liên kết.
Đột biến gen SLC2A10 gây hội chứng uốn cong động mạch làm giảm hoặc mất chức năng protein GLUT10. Thiếu protein GLUT10 khiến đường tín hiệu TGF-β hoạt động quá mức. Do đó, động mạch bị kéo dài, dẫn đến hiện tượng ngoằn ngoèo. Bên cạnh đó, tín hiệu TGF-β hoạt động quá mức cũng làm hạn chế các mô liên kết tại các bộ phận khác của cơ thể hình thành bình thường, dẫn đến các dấu hiệu và triệu chứng bổ sung của bệnh.
Chẩn đoán
Bác sĩ khám lâm sàng để xác định các triệu chứng của bệnh như xoắn động mạch, dễ chảy máu, các vị trí bị lõm hoặc lồi trên cơ thể và giãn nở động mạch. Một số xét nghiệm bổ sung nhằm đánh giá mức độ nghiêm trọng của bệnh. Siêu âm tim kiểm tra độ dày và độ đàn hồi của động mạch. Chụp cắt lớp vi tính (CT) hoặc chụp cộng hưởng từ (MRI) đánh giá tình trạng của động mạch. Chụp X-quang theo dõi tiến triển của chứng vẹo cột sống. Xét nghiệm di truyền tìm đột biến gen SLC2A10 gây bệnh.
Điều trị
Hiện nay chưa có phương pháp điều trị hoàn toàn hội chứng uốn cong động mạch. Tuy nhiên, bác sĩ sẽ chỉ định sử dụng một số loại thuốc giảm đau, antihistamin, bronchodilator làm giảm triệu chứng. Bệnh nhân cần được theo dõi chặt chẽ nhằm phát hiện và điều trị các vấn đề về tim mạch (suy tim, bệnh lý van tim) kịp thời.
Dạng di truyền
Hội chứng uốn cong động mạch di truyền theo kiểu lặn trên nhiễm sắc thể thường. Do đó, bệnh chỉ biểu hiện khi có cả hai bản sao của gen đột biến trong mỗi tế bào. Bệnh nhân mắc bệnh lặn trên nhiễm sắc thể thường sẽ có bố và mẹ mang một bản sao của gen đột biến, nhưng bố mẹ ít khi biểu hiện triệu chứng bệnh.

Nguồn: U.S. National Library of Medicine
Phòng ngừa
Bệnh di truyền lặn đột biến gen SLC2A10, cha mẹ mang đột biến dị hợp nên gần như không có biểu hiện bệnh, do đó rất khó phát hiện cho đến khi sinh con. Để chủ động phòng ngừa, cha mẹ nên làm xét nghiệm sàng lọc gen lặn để chủ động cho tương lai của con. Các cặp vợ chồng trước khi mang thai cần tư vấn và xét nghiệm di truyền đảm bảo sinh con khỏe mạnh. Người thuộc nhóm nguy cơ mắc bệnh cần khám sức khỏe và tầm soát bệnh định kỳ.
Các tên gọi khác
- Arterial tortuosity
- ATS
References
- Genetic Testing Information. Arterial tortuosity syndrome. Retrieved April 19, 2023 from https://www.ncbi.nlm.nih.gov/gtr/conditions/C1859726/
- Genetic and Rare Diseases Information Center. Arterial tortuosity syndrome. Retrieved April 19, 2023 from https://rarediseases.info.nih.gov/diseases/774/arterial-tortuosity-syndrome
- Catalog of Genes and Diseases from OMIM. ARTERIAL TORTUOSITY SYNDROME. Retrieved April 19, 2023 from https://omim.org/entry/208050
- U.S National Library of Medicine. Arterial tortuosity syndrome. Retrieved April 19, 2023 from https://medlineplus.gov/genetics/condition/arterial-tortuosity-syndrome/
- American Heart Association. Arterial Tortuosity Syndrome. Retrieved April 19, 2023 from https://www.ahajournals.org/doi/10.1161/CIRCULATIONAHA.107.739839
- Frontiers. Neonatal Arterial Tortuosity and Adult Aortic Aneurysm—Is There a Missing Link?—A Case Report. Retrieved April 19, 2023 from https://www.frontiersin.org/articles/10.3389/fped.2021.814773/full
- MalaCards. Arterial Tortuosity Syndrome (ATORS). Retrieved April 19, 2023 from https://www.malacards.org/card/arterial_tortuosity_syndrome
- National Organization for Rare Disorders. Arterial tortuosity syndrome. Retrieved April 19, 2023 from https://rarediseases.org/rare-diseases/arterial-tortuosity-syndrome/
- Orphanet. Arterial tortuosity syndrome. Retrieved April 19, 2023 from https://www.orpha.net/consor/cgi-bin/OC_Exp.php?Lng=GB&Expert=3342