Ứ mật trong gan ở trẻ sơ sinh do thiếu citrin (NICCD) là một bệnh lý về gan còn được gọi là citrullinemia loại II (Citrullinemia Type II – CIT II). NICCD ngăn chặn dòng chảy của mật (một chất dịch tiêu hóa do gan sản xuất) và cản trở cơ thể xử lý một số chất dinh dưỡng làm ứ mật trong gan dẫn đến các rối loạn chức năng gan của trẻ dưới một tuổi.
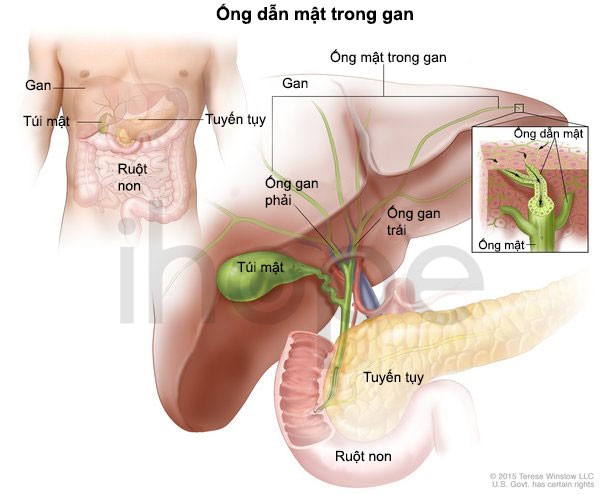
Ảnh: Ống dẫn mật trong gan
Nguồn: U.S. National Library of Medicine
Biểu hiện lâm sàng
Ứ mật trong gan ở trẻ sơ sinh do thiếu citrin (NICCD) có thể gây ra các triệu chứng như gan nhiễm mỡ, xơ gan, nhẹ cân, chậm lớn, giảm protein máu, giảm các yếu tố đông máu, thiếu máu tán huyết, gan to, rối loạn chức năng gan và hạ đường huyết ở trẻ em dưới một tuổi. NICCD thường không nghiêm trọng, các triệu chứng biến mất sau một tuổi nếu được điều trị thích hợp. Trong một số ít trường hợp, trẻ mắc bệnh phát triển các dấu hiệu và triệu chứng khác trong thời thơ ấu sau khi đã hồi phục sau NICCD, bao gồm chậm phát triển, cực kỳ mệt mỏi và lipid bất thường trong máu (rối loạn lipid máu). Đây tình trạng không phát triển và rối loạn lipid máu do thiếu citrin (FTTDCD). Vào khoảng hai tuổi, trẻ bị NICCD có xu hướng thích ăn các loại thực phẩm giàu đạm và chất béo cũng như ác cảm với các loại thực phẩm có đường và giàu carbohydrate. Khi lớn lên, một số người có thể phát triển các triệu chứng liên quan đến hệ thần kinh như lú lẫn, bồn chồn, mất trí nhớ, hành vi bất thường (hung hăng, cáu kỉnh và tăng động), co giật và hôn mê. Đây là các triệu chứng đặc trưng của bệnh citrullinemia loại II khởi phát ở người lớn (CTLN2).
Độ phổ biến
Ước tính tỉ lệ trẻ mắc bệnh là 1/19.000 tại Nhật Bản, 1/50.000 tại Hàn Quốc và 1/17.000 tại Trung Quốc. Trong đó, tỉ lệ mang gen đột biến gây bệnh chiếm 1/65 tại Nhật Bản, chiếm 1/65 tại Trung Quốc, chiếm 1/48 tại khu vực miền nam Trung Quốc bao gồm Đài Loan và chiếm 1/112 tại Hàn Quốc. Thiếu men citrin cũng được ghi nhận tại một số quốc gia khác như Israel, Pakistan, Hoa Kỳ, Vương quốc Anh và Cộng hòa Séc.
Nguyên nhân
Các đột biến gen SLC25A13 gây ra tình trạng ứ mật trong gan của trẻ sơ sinh do thiếu citrin cũng như bệnh citrullinemia loại II khởi phát trên người trưởng thành. Gen này cung cấp hướng dẫn để tạo ra một loại protein gọi là citrin. Trong tế bào, citrin vận chuyển các phân tử nhằm sản xuất và phân hủy đường đơn, sản xuất protein và chu trình urê. Các phân tử được vận chuyển bởi citrin cũng tham gia vào quá trình tạo ra nucleotide—khối cấu tạo nên ADN và ARN. Các đột biến gen SLC25A13 thường ngăn cản tế bào tạo ra citrin có chức năng, nên chu trình urê bị ức chế cũng như quá trình sản xuất protein và nucleotide bị gián đoạn. Do đó, amoniac và các chất độc hại khác tích tụ đến mức gây ra các dấu hiệu và triệu chứng của bệnh ứ mật trong gan trên trẻ sơ sinh. Thiếu citrin cũng dẫn đến các triệu chứng của citrullinemia loại II khởi phát trên người trưởng thành.
Chẩn đoán
Chẩn đoán thiếu citrin được xác định thông qua kết quả xét nghiệm sinh hóa phù hợp với tình trạng thiếu citrin và các biến thể gây bệnh trên gen SLC25A13 bằng xét nghiệm di truyền. Phân tích Western blot có thể được thực hiện nếu không xác định được các biến thể gây bệnh.
Đối với trẻ sơ sinh, trong vòng 24–48 giờ sau khi chào đời, trẻ được lấy máu gót chân để thực hiện sàng lọc sơ sinh nhằm phát hiện rối loạn chuyển hóa citrulline loại II và các bệnh lý khác. Nếu kết quả cho thấy nguy cơ cao, bác sĩ sẽ cho em bé làm thêm xét nghiệm chẩn đoán. Điều quan trọng cần lưu ý là kết quả sàng lọc nguy cơ cao không đồng nghĩa với kết luận em bé mắc bệnh. Kết quả này có thể do mẫu máu ban đầu được thu quá ít hoặc quá sớm. Tuy nhiên, cha mẹ nên đưa trẻ tái khám để làm xét nghiệm xác nhận. Nếu không được điều trị, rối loạn chuyển hóa citrulline loại II có thể ảnh hưởng đến sức khỏe của trẻ rõ rệt ngay sau khi sinh, xét nghiệm tiếp theo phải được tiến hành càng sớm càng tốt để xác định xem liệu trẻ có mắc bệnh hay không.
Xét nghiệm tiếp theo bao gồm kiểm tra mẫu nước tiểu và máu của em bé để tìm dấu hiệu của rối loạn chuyển hóa citrullin. Một số chất có hại tích tụ với nồng độ cao trong cơ thể khi trẻ bị rối loạn axit amin, do đó, bác sĩ có thể dựa vào nồng độ các chất này để xác định xem bé có mắc bệnh hay không. Nồng độ citrulline cao trong máu có thể cho thấy em bé bị rối loạn chuyển hóa citrulline II. Đôi khi, bác sĩ cần xét nghiệm thêm một mẫu da rất nhỏ.
Ngoài ra, bác sĩ có thể chỉ định xét nghiệm gen nhằm phát hiện đột biến gây bệnh, từ đó kết quả chẩn đoán được xác nhận.
Xét nghiệm sinh hóa
Quá trình chẩn đoán thiếu citrin được hỗ trợ thêm bởi kết quả của xét nghiệm sau:
- Amoniac
- Phân tích định lượng axit amin
- Chất ức chế trypsin bài tiết tuyến tụy (PSTI)
Xét nghiệm di truyền
Các phương pháp tiếp cận xét nghiệm di truyền phân tử có thể bao gồm xét nghiệm đơn gen và đa gen.
- Xét nghiệm đơn gen: phân tích trình tự của gen SLC25A13 và xác định điểm đột biến trên gen.
- Xét nghiệm đa gen: phân tích gen SLC25A13 và các gen liên quan.
Phân tích Western Blot
Western blot là kỹ thuật phân tích được sử dụng rộng rãi trong sinh học phân tử và di truyền miễn dịch để phát hiện các protein cụ thể trong một mẫu mô đồng nhất hoặc chiết xuất.
Phân tích Western blot đối với protein citrin có thể được thực hiện khi cả hai loại xét nghiệm trên không cho thấy sự thiếu hụt citrin và không phát hiện biến thể gây bệnh trên gen SLC25A13.
Điều trị
Điều trị cho trẻ ứ mật do thiếu men citrin bao gồm chế độ ăn uống bổ sung các loại vitamin tan trong dầu và sữa công thức không chứa lactose (ở trẻ có biểu hiện tăng bài tiết galactose) và bổ sung chất béo MCT (medium chain triglycerides). Hạn chế lactose làm giảm galactosemia, sử dụng MCT cải thiện tình trạng ứ mật. Thiếu hụt citrin dường như là sự thiếu hụt năng lượng ở gan do những thay đổi trong chức năng chuyển hóa của quá trình đường phân. MCT được thủy phân và hấp thụ dưới dạng axit béo chuỗi trung bình tự do để cung cấp ATP trực tiếp cho gan.
Ngoài ra, để ngăn chặn sự khởi phát của CTLN2 hoặc FTTDCD làm trầm trọng thêm các triệu chứng, cần thực hiện:
- Chế độ ăn giàu protein và lipid, ít carbohydrate
- Bổ sung chất béo MCT có thể giảm nguy cơ hạ đường huyết và giảm mức amoniac trong máu vì nó cải thiện tình trạng thiếu ATP ở gan.
- Dùng arginine và natri pyruvate có thể ngăn ngừa các cơn tăng huyết áp.
Những điều cần tránh đối với bệnh nhân thiếu citrin:
- Chế độ ăn ít protein, carbohydrate cao, calo cao có thể ngăn ngừa chứng tăng urê huyết do thiếu hụt enzyme chu trình urê, tuy nhiên có thể gây hại cho những người bị thiếu citrin ở bất kỳ dạng bệnh nào (ví dụ, NICCD, FTTDCD hoặc CTLN2). Chế độ ăn nhiều carbohydrate tăng sản xuất NADH, làm gián đoạn quá trình tổng hợp urê và kích thích quá trình vận chuyển citrate-malate, có thể dẫn đến tăng đường huyết, gan nhiễm mỡ và tăng triglycerid máu.
- Truyền đường như glycerol, fructose và glucose. Bệnh nhân CTLN2 bị phù não nặng, được điều trị bằng thuốc chứa glycerol vẫn tiếp tục xấu đi. Do đó, chống chỉ định sử dụng glycerol đối với những người mắc CTLN2. NADH được tạo ra khi phân hủy một lượng lớn glycerol và fructose trong tế bào gan. Tác dụng này có thể làm rối loạn chức năng gan và tạo ra các chất độc hại. Tình trạng hạ đường huyết cũng trở nên tồi tệ hơn khi truyền glucose nồng độ cao. Lưu ý: truyền mannitol dường như an toàn hơn.
- Rượu. Alcohol dehydrogenase (ADH) tạo ra NADH trong tế bào gan, do đó, uống rượu có thể gây khởi phát CTLN2. Rượu cần được tránh tuyệt đối.
- Thuốc. Acetaminophen (paracetamol) và rabeprozole có thể gây ra CTLN2.
Dạng di truyền
Ứ mật do thiếu men citrin được di truyền theo kiểu lặn trên nhiễm sắc thể thường, nghĩa là cả hai bản sao của gen tương ứng trong mỗi tế bào đều có đột biến. Cha mẹ của những người mắc bệnh mang một bản sao của gen đột biến, nhưng họ thường không biểu hiện các dấu hiệu và triệu chứng của bệnh.

Nguồn: U.S. National Library of Medicine
Phòng ngừa
Ứ mật do thiếu men citrin thường được di truyền theo gen lặn, nên bệnh rất khó phát hiện với những người thể mang vì họ gần như không có biểu hiện rõ ràng, đến khi có con mới biết được thì đã quá muộn. Do đó, các cặp vợ chồng trước khi mang thai cần làm xét nghiệm gen lặn để sàng lọc căn bệnh này nhằm đảm bảo sinh con khỏe mạnh và lành lặn.
Các tên gọi khác
- CIT
- Citrullinuria
- Citrin deficiency
- CTLN II
- Neonatal intrahepatic cholestasis caused by citrin deficiency (neonatal form only)
- NICCD (neonatal form only)
- Citrullinemia
References
- Genetic Testing Information. Neonatal intrahepatic cholestasis caused by citrin deficiency. Retrieved September 25, 2021 from https://www.ncbi.nlm.nih.gov/gtr/conditions/C1853942/
- Genetic and Rare Diseases Information Center. Citrullinemia type I. Retrieved September 21, 2021 from https://rarediseases.info.nih.gov/diseases/6114/citrullinemia-type-i
- Catalog of Genes and Diseases from OMIM. CITRULLINEMIA, TYPE II, NEONATAL-ONSET. Retrieved September 21, 2021 from https://omim.org/entry/605814
- U.S National Library of Medicine. Citrullinemia. Retrieved September 19, 2021 from https://medlineplus.gov/genetics/condition/citrullinemia
- National Center for Biotechnology Information. Citrin Deficiency. Retrieved September 19, 2021 from https://www.ncbi.nlm.nih.gov/books/NBK1181/
- Citrin Foundation. HOW TO MONITOR FOR CITRIN DEFICIENCY. Retrieved September 19, 2021 from https://citrinfoundation.org/what-is-citrin-deficiency/monitoring-treatments/
- National Center for Advancing Translational Sciences. Neonatal intrahepatic cholestasis caused by citrin deficiency Retrieved September 19, 2021 from https://rarediseases.info.nih.gov/diseases/10214/neonatal-intrahepatic-cholestasis-caused-by-citrin-deficiency
- Citrin Foundation. Prevention & Precautions. Retrieved September 19, 2021 from https://citrinfoundation.org/what-is-citrin-deficiency/prevention-precautions/