Xơ tủy nguyên phát gây ra các mô sẹo (xơ hóa) tích tụ trong tủy xương. Do xơ hóa, tủy xương không thể sản sinh đủ các tế bào máu bình thường, từ đó gây ra các dấu hiệu và triệu chứng của bệnh.
Nguồn: Medlineplus.gov
Biểu hiện lâm sàng
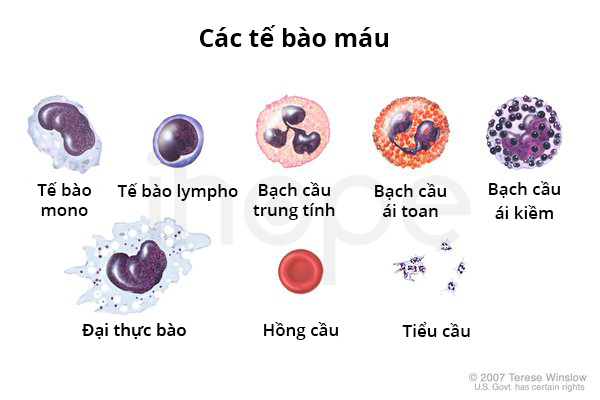
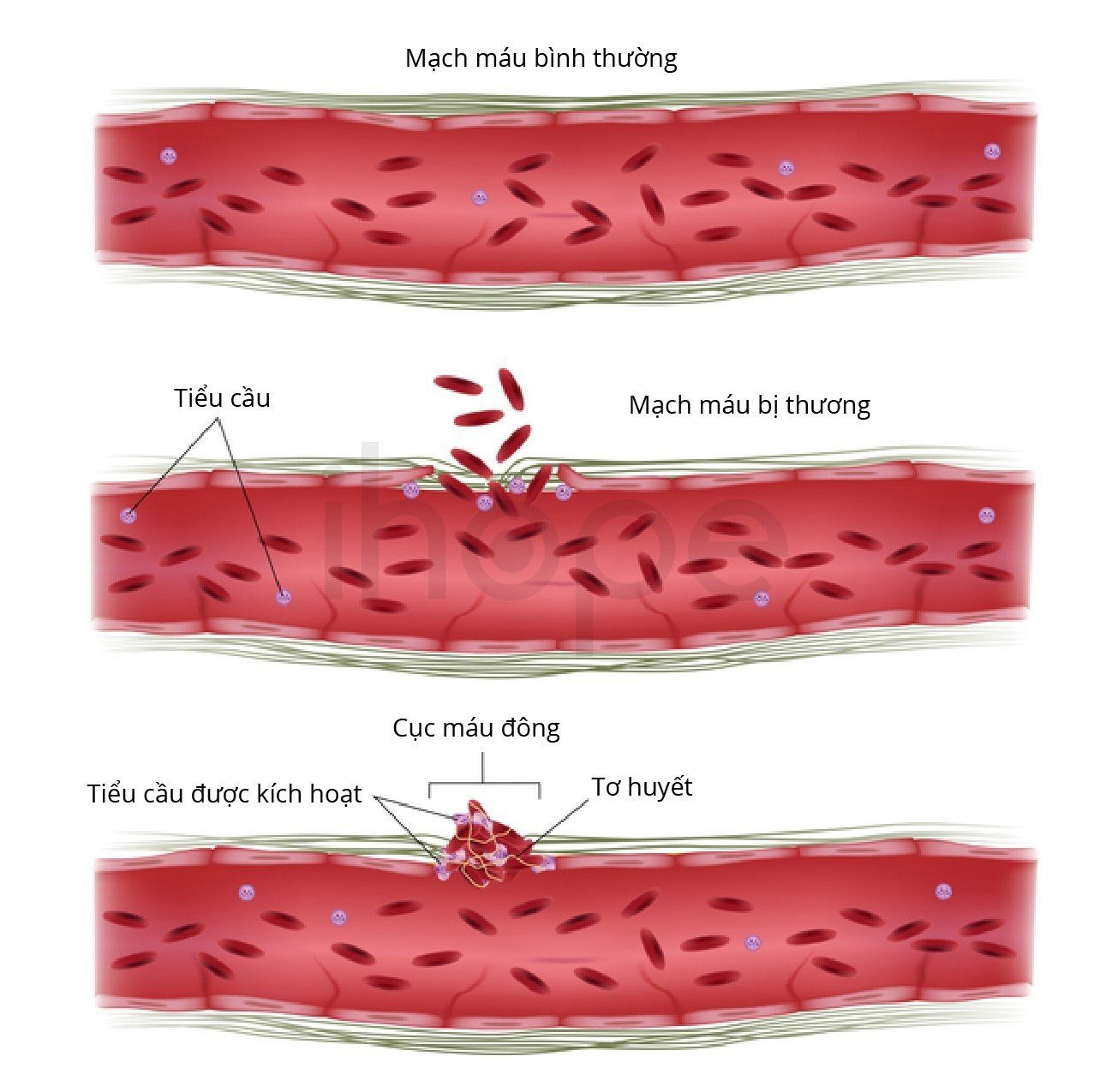
Trong phần lớn trường hợp, giai đoạn đầu của bệnh xơ tủy nguyên phát không gây ra triệu chứng. Quá trình xơ hóa phát triển theo thời gian cho đến khi số lượng các tế bào máu sụt giảm. Thiếu hồng cầu khiến bệnh nhân mệt mỏi và khó thở. Người bệnh không tạo ra đủ lượng bạch cầu dẫn đến tăng nguy cơ nhiễm trùng. Số lượng tiểu cầu giảm gây ra tình trạng chảy máu hoặc đông máu bất thường và các vết bầm tím trên da.
Nguồn: Medlineplus.gov
Quá trình tạo máu trong tủy xương bị gián đoạn nên các cơ quan khác như gan và lách tăng cường sản xuất tế bào máu (tạo máu ngoài tủy). Do đó, gan và lách trở nên to hơn. Bệnh nhân phát triển chứng lách to cảm thấy đau và đầy bụng, chủ yếu bên dưới mạn sườn trái. Ngoài ra, người mắc xơ tủy nguyên phát có thể bị sốt, đổ mồ hôi ban đêm và nhức xương.
Xơ tủy nguyên phát xảy ra tại mọi lứa tuổi nhưng đa phần bệnh phát triển trong giai đoạn từ 50 đến 80 tuổi.
Độ phổ biến
Người ta ước tính tỷ lệ mắc xơ tủy nguyên phát khoảng 1/500.000 người trên toàn thế giới.
Nguyên nhân
Đột biến một trong những gen JAK2, MPL, CALR và TET2 gây ra phần lớn các trường hợp xơ tủy nguyên phát. Gen JAK2 và MPL cung cấp hướng dẫn tạo ra các protein thúc đẩy quá trình phát triển và phân chia tế bào máu. Gen CALR cung cấp các hướng dẫn tạo ra một loại protein có chưc năng duy trì nồng độ canxi ổn định trong tế bào và đảm bảo các protein gấp cuộn đúng. Protein từ gen TET2 vẫn chưa rõ chức năng.
Protein tạo ra từ gen JAK2 và MPL phối hợp với nhau nhằm kích hoạt đường truyền tín hiệu hóa học JAK/STA từ bên ngoài vào bên trong nhân tế bào. Tín hiệu này thúc đẩy quá trình tăng sinh các tế bào máu, chủ yếu tiểu cầu và megakaryocyte (tiền thân tiểu cầu).
Đột biến gen JAK2, MPL khiến đường tín hiệu JAK/STAT hoạt động quá mức dẫn đến sản xuất dư thừa megakaryocyte. Do đó, tế bào được kích thích sản sinh và giải phóng collagen—protein hỗ trợ cấu trúc tế bào. Lượng protein này dư thừa tích tụ đến mức hình thành mô sẹo trong tủy xương.
Những trường hợp mắc bệnh mang đột biến gen CALR và TET2 đã được ghi nhận nhưng cơ chế chưa rõ ràng. Một số ít bệnh nhân không có các đột biến gen liên quan.
Chẩn đoán
Tăng tiểu cầu tiên phát được chẩn đoán dựa trên biểu hiện lâm sàng, tiền sử bệnh trong gia đình. Một số xét nghiệm chuyên biệt giúp chẩn đoán bệnh bao gồm:
- Công thức máu toàn phần (complete blood count) cho phép định lượng tất cả các tế bào máu.
- Phết máu ngoại biên dưới kính hiển vi nhằm quan sát hình dạng và kích thước tế bào máu.
- Xét nghiệm tủy xương để chọc hút hoặc sinh thiết tủy xương và kiểm tra các bất thường.
Ngoài ra, xét nghiệm di truyền nhằm phát hiện các đột biến gen liên quan là phương pháp chẩn đoán nhanh và chính xác.
Điều trị
Hiện nay chưa có phương pháp điều trị dứt điểm xơ tủy nguyên phát. Các liệu pháp giúp giảm nhẹ các triệu chứng nhằm cải thiện chất lượng đời sống bệnh nhân.
Một số loại thuốc có thể được chỉ định bao gồm:
- Jakafi, Inrebic và Vonjo: được FDA phê duyệt nhằm điều trị xơ tủy từ nhẹ đến trung bình.
- Androgens: tăng cường sản sinh hồng cầu.
- Thuốc điều hòa hệ miễn dịch (immunomodulators), hóa trị: giúp giảm nhẹ các biểu hiện bệnh.
Trường hợp mắc bệnh bị thiếu máu nghiêm trọng, truyền máu có thể được chỉ định.
Dạng di truyền
Xơ tủy nguyên phát thường không di truyền. Bệnh phát sinh từ đột biến gen xảy ra trong các tế bào tạo máu sớm sau khi thụ thai. Những bất thường này được gọi là đột biến soma.
Phòng ngừa
Hiện nay chưa có phương pháp phòng ngừa bệnh xơ tủy nguyên phát.
Các tên gọi khác
- Agnogenic myeloid metaplasia
- Chronic idiopathic myelofibrosis
- Idiopathic myelofibrosis
- Myelofibrosis with myeloid metaplasia
- Myeloid metaplasia
References
- Genetic Testing Information. Primary myelofibrosis. Retrieved 13 June 2023 from https://www.ncbi.nlm.nih.gov/gtr/conditions/C0001815/?_ga=2.102206725.546406511.1686539465-1635318420.1684225451 Genetic Testing Information. Primary myelofibrosis. Retrieved 13 June 2023 from https://www.ncbi.nlm.nih.gov/gtr/conditions/C0001815/?_ga=2.102206725.546406511.1686539465-1635318420.1684225451
- Genetic and Rare Diseases Information Center. Primary myelofibrosis. Retrieved 13 June 2023 from https://rarediseases.info.nih.gov/diseases/8618/primary-myelofibrosis
- Catalog of Genes and Diseases from OMIM. MYELOFIBROSIS WITH MYELOID METAPLASIA, INCLUDED; MMM, INCLUDED. Retrieved 13 June 2023 from https://omim.org/entry/254450
- U.S National Library of Medicine. Primary myelofibrosis. Retrieved 13 June 2023 from https://medlineplus.gov/genetics/condition/primary-myelofibrosis/#inheritance
- Frontiers. Pediatric immune myelofibrosis (PedIMF) as a novel and distinct clinical pathological entity. Retrieved 13 June 2023 from https://www.frontiersin.org/articles/10.3389/fped.2022.1031687/full
- Mayo Clinic. Myelofibrosis. Retrieved 13 June 2023 from https://www.mayoclinic.org/diseases-conditions/myelofibrosis/symptoms-causes/syc-20355057
- MSD Manuals. Primary Myelofibrosis. Retrieved 13 June 2023 from https://www.msdmanuals.com/professional/hematology-and-oncology/myeloproliferative-disorders/primary-myelofibrosis
- National Organization for Rare Disorders. Primary Myelofibrosis. Retrieved 13 June 2023 from https://rarediseases.org/rare-diseases/primary-myelofibrosis/
- Orphanet. Primary myelofibrosis. Retrieved 13 June 2023 from https://www.orpha.net/consor/cgi-bin/OC_Exp.php?Lng=GB&Expert=824