
Hội chứng mất đoạn 8p23.1 (8p23.1 microdeletion syndrome) bắt nguồn từ mất đoạn trên nhiễm sắc thể số 8. Hội chứng này gây ra một loạt các bất thường liên quan đến phát triển thể chất và trí tuệ. Nguyên nhân chủ yếu do các gen quan trọng bị mất trên vùng nhiễm sắc thể này.
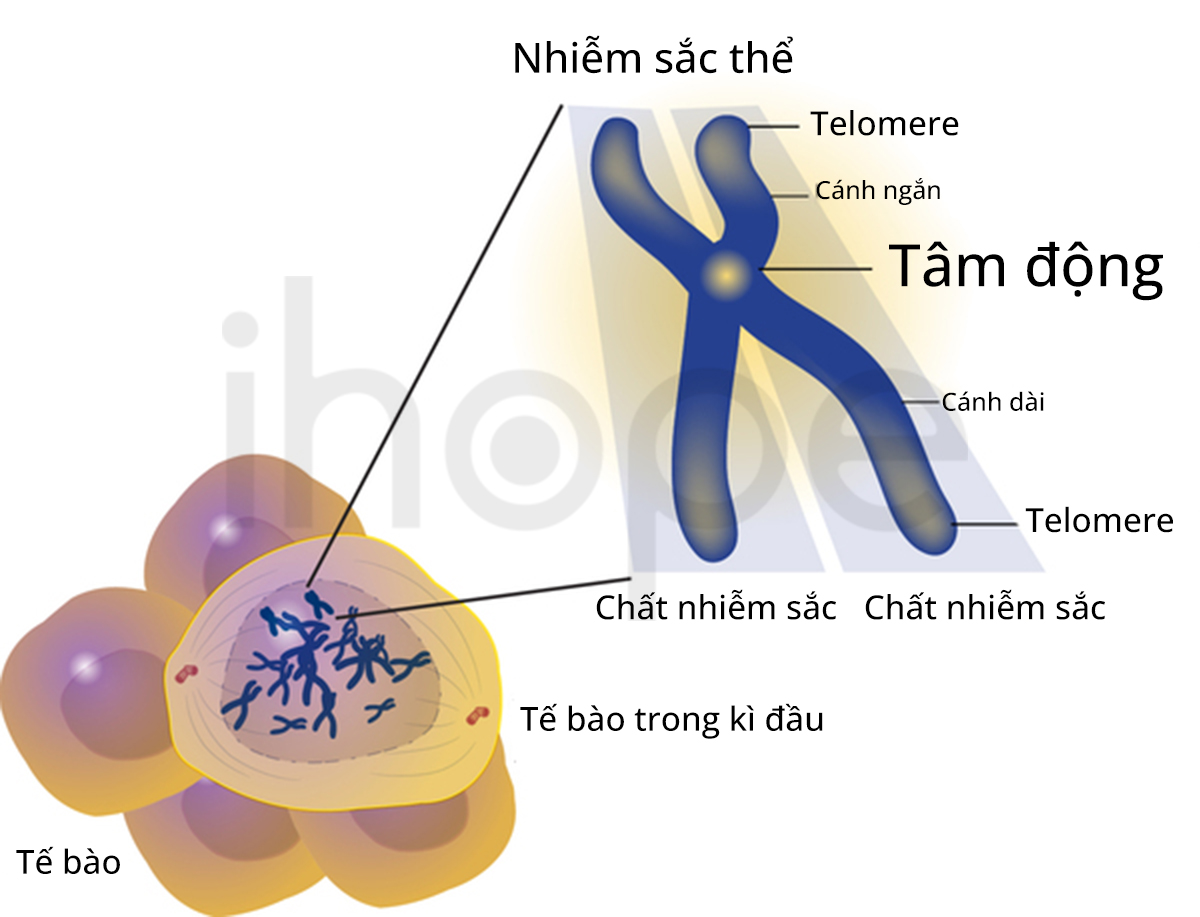
Nguồn: U.S. National Library of Medicine
Biểu hiện lâm sàng
Biểu hiện lâm sàng của hội chứng mất đoạn 8p23.1 rất đa dạng và không phụ thuộc vào kích thước đoạn mất. Mức độ nghiêm trọng cũng khác nhau giữa các cá nhân. Một số người bệnh có triệu chứng nhẹ, họ có thể đạt chỉ số thông minh bình thường sau khi điều trị chậm phát triển vận động và ngôn ngữ.
Các triệu chứng phổ biến bao gồm:
- Tăng trưởng chậm trước và sau khi sinh
- Cân nặng lúc sinh thấp
- Chậm phát triển trí tuệ và ngôn ngữ
Nhiều trường hợp, trẻ có các dấu hiệu bất thường về hành vi như tăng động. Một số trẻ xuất hiện bệnh lí về tim và thoát vị cơ hoành.
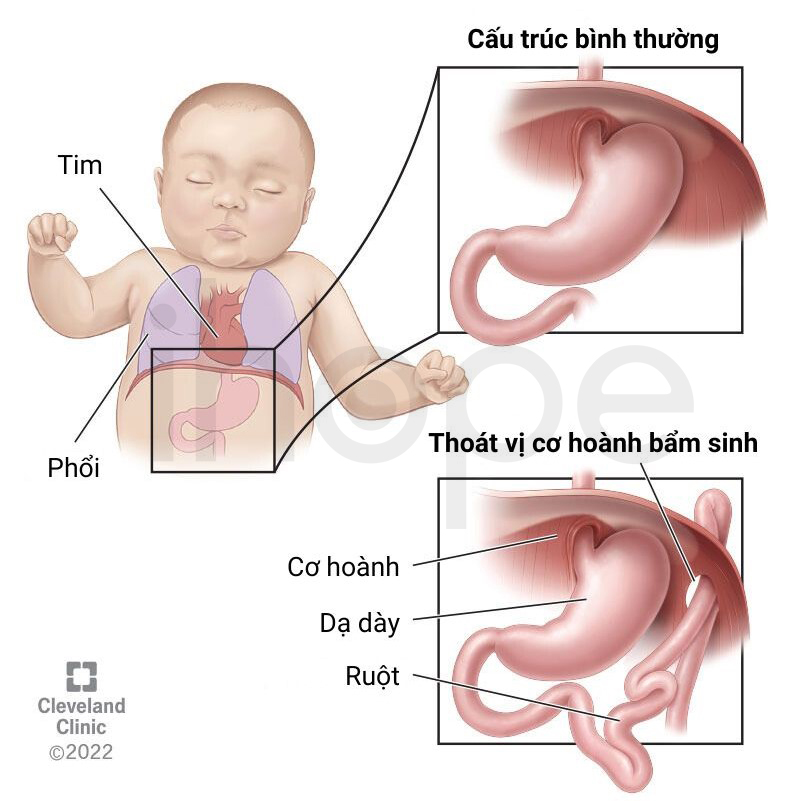
Nguồn: Cleveland Clinic
Bệnh lí về tim do hội chứng mất đoạn 8p23.1 gây ra có thể ảnh hưởng đến tuổi thọ của người bệnh. Phần lớn những bất thường về tim này không quá nghiêm trọng và bệnh nhân có thể điều trị bằng phẫu thuật. Tuy nhiên, trong một số trường hợp, biến chứng tim mạch hoặc hô hấp với mức độ nghiêm trọng có thể dẫn đến tử vong cho trẻ trong giai đoạn sơ sinh.
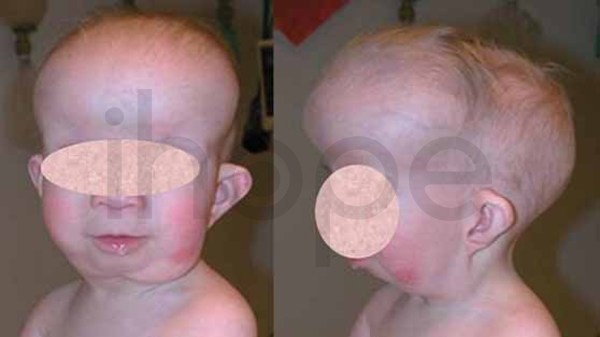
Ngoài ra, người bệnh có các điểm dị biệt trên khuôn mặt như:
- Đầu nhỏ
- Trán gồ
- Mũi lớn và tẹt
- Tai dị dạng

Ảnh: Tật đầu nhỏ
Nguồn: Centers for Disease Control and Prevention
Ảnh: Trán nhô ra phía trước
Nguồn: Elements of Morphology, National Human Genome Research Institute
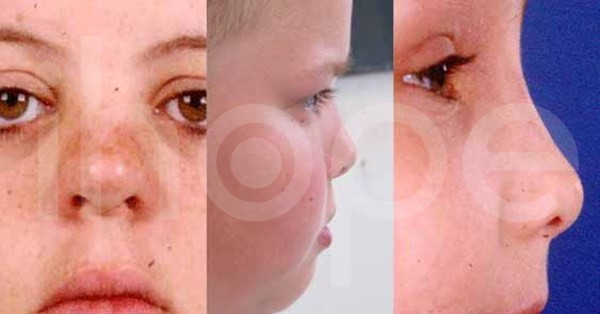
Ảnh: Mũi thấp, ngắn và hếch
Nguồn: Elements of Morphology, National Human Genome Research Institute
Độ phổ biến
Hội chứng mất đoạn 8p23.1 là bệnh di truyền hiếm gặp. Hiện này, hơn 50 trường hợp mắc bệnh trên toàn thế giới đã được ghi nhận.
Nguyên nhân
Bất thường trong quá trình sao chép ADN trên các vùng lặp lại của nhiễm sắc thể 8 là nguyên nhân chủ yếu gây ra hội chứng mất đoạn 8p23.1. Đoạn mất có kích thước khoảng 3,4 Mb và mang nhiều gen quan trọng như GATA4, SOX7, TNKS và MCPH1.
Gen GATA4 có chức năng quan trọng đối với quá trình phát triển tim. Thiếu một bản sao của gen này ảnh hưởng đến cấu trúc của tim hoặc gây ra khiếm khuyết vách tâm nhĩ, nên nhiều bệnh lí về tim bẩm sinh biểu hiện. Vì vậy, nhiều bệnh nhân mắc hội chứng mất đoạn 8p23.1 có các bất thường về tim mạch.
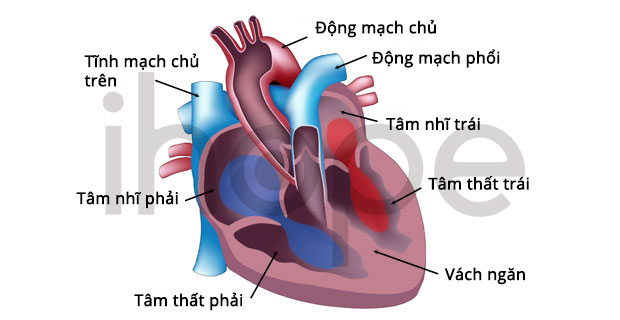
Nguồn: American Heart Association
Ngoài ra, đột biến mất các gen SOX7, TNKS và MCPH1 cũng góp phần gây ra những triệu chứng khác. Gen SOX7 tham gia nhiều hoạt động trong cơ thể, mặt khác, gen TNKS và MCPH1 liên quan đến quá trình phát triển thần kinh. Do đó, thiếu các gen này gây ra tật đầu nhỏ, người bệnh gặp vấn đề với nhận thức và phát triển thần kinh.
Chẩn đoán
Hội chứng mất đoạn 8p23.1 được chẩn đoán dựa trên đánh giá biểu hiện lâm sàng kết hợp với xét nghiệm di truyền. Phương pháp nhiễm sắc thể đồ không phát hiện được các mất đoạn nhỏ. Do đó, người ta ưu tiên sử dụng kĩ thuật hiện đại như nhiễm sắc thể đồ phân tử (molecular karyotyping) kết hợp với các phương pháp như lai huỳnh quang tại chỗ (FISH), khuếch đại đầu dò phụ thuộc ligase (MLPA) hoặc so sánh bộ gen trên mảng (aCGH) nhằm xác định chính xác đặc điểm di truyền của đoạn mất.
Đối với chẩn đoán trước sinh, thai phụ có thể thực hiện chọc ối hoặc sinh thiết gai nhau kết hợp với phân tích di truyền tế bào. Chẩn đoán sớm cho phép bác sĩ can thiệp kịp thời và tối ưu hóa kết quả điều trị. Trẻ mắc bệnh có thể cải thiện đáng kể tiên lượng và chất lượng cuộc sống nếu được điều trị ngay từ giai đoạn đầu.
Điều trị
Hiện nay, chưa có phương pháp điều trị hoàn toàn hội chứng mất đoạn 8p23.1. Các liệu pháp chủ yếu nhằm giảm nhẹ triệu chứng và cải thiện chất lượng đời sống người bệnh.
Dạng di truyền
Phần lớn các trường hợp mắc hội chứng mất đoạn 8p23.1 không di truyền. Bệnh xảy ra do đột biến mới (de novo) xuất hiện trong quá trình hình thành tế bào sinh sản (trứng, tinh trùng) hoặc quá trình phát triển phôi sớm. Các trường hợp di truyền bệnh từ cha mẹ hiếm gặp hơn.
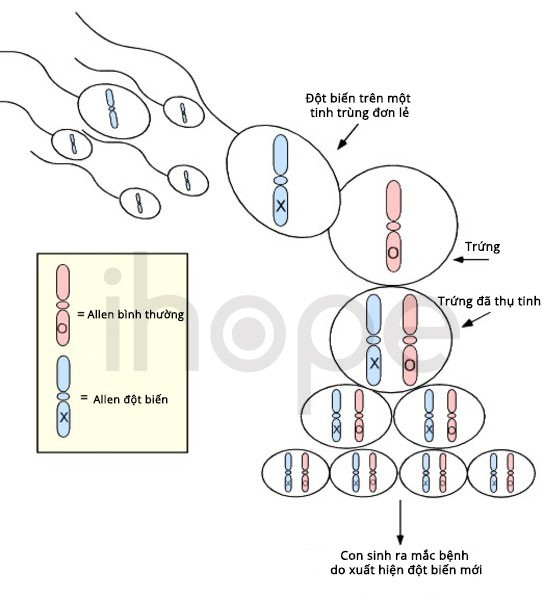
Nguồn: U.S. National Library of Medicine
Trong một số trường hợp, bệnh di truyền từ bố mẹ do hiện tượng chuyển đoạn không cân bằng. Hiện tượng này xảy ra khi hoạt động trao đổi đoạn giữa các nhiễm sắc thể không tương đồng, dẫn đến bất thường cấu trúc nhiễm sắc thể. Trẻ nhận các bất thường này có thể bị rối loạn biểu hiện gen và gây ra biểu hiện bệnh.
Phòng ngừa
Hội chứng mất đoạn 8p23.1 do đột biến ngẫu nhiên xảy ra trong quá trình tạo phôi, vì vậy tất cả thai phụ đều có nguy cơ mang thai bị bệnh. Do đó, sàng lọc phát hiện bệnh sớm vô cùng quan trọng và cần thiết. Sản phụ nên thăm khám và siêu âm định kì cũng như làm các xét nghiệm cần thiết như sàng lọc NIPT nhằm phát hiện sớm vấn đề có thể xảy ra với thai nhi.
Các tên gọi khác
- Del(8)(p23.1)
- Monosomy 8p23.1
References
- Genetic Testing Information. 8p23.1 microdeletion syndrome. Retrieved November 18, 2024 from https://www.ncbi.nlm.nih.gov/gtr/conditions/C2931638/
- Genetic and Rare Diseases Information Center. 8p23.1 microdeletion syndrome. Retrieved November 18, 2024 from https://rarediseases.info.nih.gov/diseases/3769/8p231-microdeletion-syndrome
- National Institute of Health. Expanding the Phenotype of 8p23.1 Deletion Syndrome: Eight New Cases Resembling the Clinical Spectrum of 22q11.2 Microdeletion. Retrieved November 18, 2024 from https://pubmed.ncbi.nlm.nih.gov/36067875/
- National Institute of Health. Microarray Analysis of 8p23.1 Deletion in New Patients with Atypical Phenotypical Traits. Retrieved November 18, 2024 from https://pmc.ncbi.nlm.nih.gov/articles/PMC4906530/
- National Organization for Rare Disorders. 8p23.1 microdeletion syndrome. Retrieved November 18, 2024 from https://rarediseases.org/mondo-disease/8p23-1-microdeletion-syndrome/
- Orphanet. 8p23.1 microdeletion syndrome. Retrieved November 18, 2024 from https://www.orpha.net/en/disease/detail/251071





