
Thất điều với mất vận động nhãn cầu (ataxia with oculomotor apraxia) khiến cơ thể mất khả năng giữ thăng bằng và hạn chế điều khiển nhãn cầu . Theo thời gian, bệnh tiến triển nghiêm trọng hơn với các vấn đề vận động. Bệnh được chia thành nhiều nhóm, trong đó phổ biến nhất là nhóm 1, 2 và 4 có triệu chứng tương tự nhau nhưng do đột biến gen khác nhau gây ra.
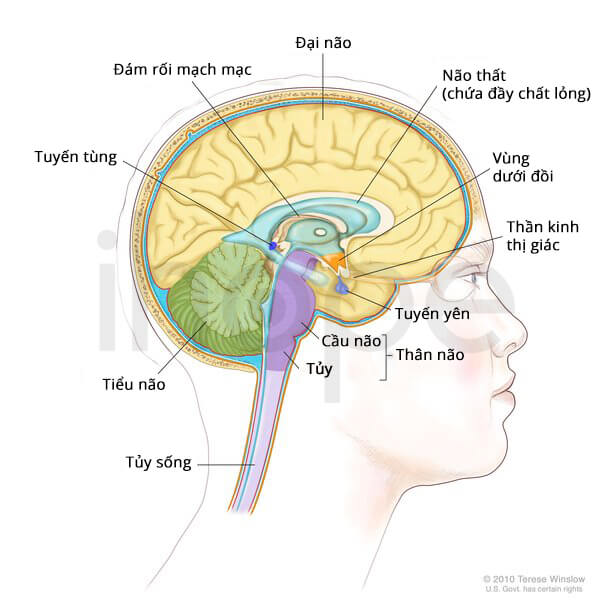
Nguồn: Terese Winslow
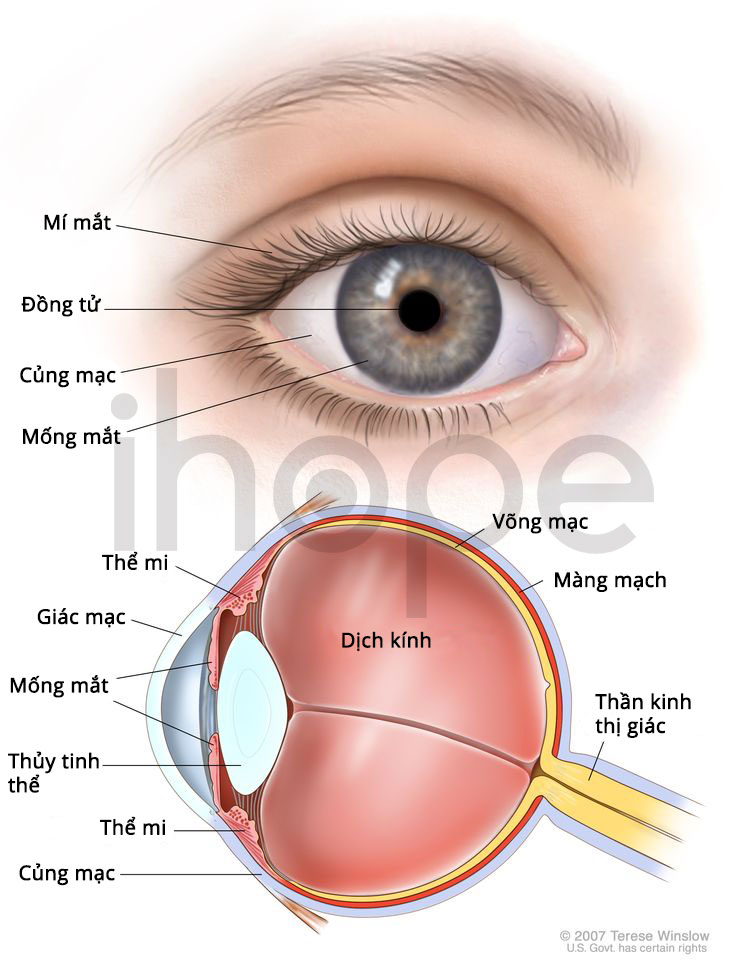
Ảnh: Cấu trúc của mắt
Nguồn: Medlineplus.gov
Biểu hiện lâm sàng
Dấu hiệu đầu tiên và phổ biến nhất của người mắc bệnh thất điều với mất vận động nhãn cầu là khả năng phối hợp và giữ thăng bằng kém. Bên cạnh đó, người bệnh cũng khó di chuyển nhãn cầu mắt từ hướng này sang hướng kia để mở rộng tầm nhìn. Mặc dù bệnh không ảnh hưởng đến trí não nhưng một vài bệnh nhân bị chậm phát triển trí tuệ.
Thất điều với mất vận động nhãn cầu nhóm 1
Thất điều với mất vận động nhãn cầu nhóm 1 thường khởi phát khi người bệnh khoảng 4 tuổi với biểu hiện như:
- Chứng múa giật
- Động kinh
- Teo cơ tay và chân
- Bệnh lý về thần kinh
Theo thời gian, những triệu chứng bệnh liên quan đến khả năng vận động có xu hướng giảm dần. Tuy nhiên, người bệnh vẫn bị ảnh hưởng đến cử động các chi, mất khả năng cảm nhận rung động và phản xạ chậm. Nhiều bệnh nhân phải ngồi xe lăn sau 10 đến 15 năm phát bệnh. Ngoài ra, bệnh nhân thuộc nhóm 1 có thể bị giảm lượng protein albumin—protein vận chuyển các phân tử trong máu. Thiếu albumin dẫn đến nồng độ cholesterol trong máu tăng cao, từ đó người bệnh tăng nguy cơ mắc bệnh tim.
Thất điều và mất vận động nhãn cầu nhóm 2
Trường hợp người mắc thất điều với mất vận động nhãn cầu nhóm 2 khởi phát bệnh từ khoảng 15 tuổi với biểu hiện tương tự nhóm 1 là chứng múa giật và động kinh. Tuy nhiên, triệu chứng này trở thành mãn tính và không thuyên giảm theo thời gian. Người bệnh cũng gặp thêm vấn đề thần kinh.
Người bệnh thuộc nhóm 2 hàm lượng cao hai loại protein sau:
- Alpha-fetoprotein (AFP): thường hiện diện trong máu phụ nữ mang thai
- Creatine phosphokinase (CPK) có mặt chủ yếu trong mô cơ
Người ta vẫn chưa rõ nồng độ cao hai loại protein này tác động thế nào lên cơ thể bệnh nhân. Tuy người bệnh nhóm 2 có mức albumin bình thường nhưng nồng độ cholesterol có thể tăng cao.

Ảnh: Các loại mô cơ
Nguồn: Medlineplus.gov
Thất điều và mất vận động nhãn cầu nhóm 4
Đối với người mắc thất điều với mất vận động nhãn cầu thuộc nhóm 4, bệnh khởi phát vào khoảng 4 tuổi. Bên cạnh những biểu hiện lâm sàng, người bệnh thuộc nhóm 4 có khả năng tiến triển chứng loạn trương lực cơ nhưng chúng có xu hướng tự khỏi. Phần lớn bệnh nhân có hàm lượng albumin, nồng độ cholesterol và alpha-fetoprotein trong máu bình thường, nhưng có một số bệnh nhân bị ảnh hưởng.
Biểu hiện khác của người bệnh thuộc nhóm 4 bao gồm:
- Căng cơ kéo dài
- Teo cơ tay và chân
- Bệnh lý thần kinh
Độ phổ biến
Thất điều với mất chuyển động nhãn cầu là bệnh di truyền hiếm gặp. Người bệnh thuộc nhóm 1 và 4 phổ biến tại Bồ Đào Nha. Nhóm 1 cũng được ghi nhận nhiều tại Nhật Bản. Người ta ước tính tỉ lệ mắc bệnh nhóm 2 là 1/900.000 trường hợp trên thế giới.
Nguyên nhân
Đột biến gen APTX, SETX hoặc PNKP gây ra thất điều và mất chuyển động nhãn cầu nhóm 1, 2 hoặc 4 tương ứng. Các gen APTX, SETX và PNKP cung cấp hướng dẫn tạo ra protein liên quan đến sửa chữa ADN sai hỏng. Đột biến những gen này khiến lượng protein chức năng giảm số lượng, dẫn đến quá trình sửa sai không hiệu quả và chuỗi ADN đứt gãy tích tụ.
Thông thường, ADN bị phá hủy do gốc oxy hóa tự do được tạo ra từ hoạt động của tế bào hoặc khi tế bào tiếp xúc với bức xạ hoặc môi trường ô nhiễm. ADN cũng có nguy cơ đứt gãy khi nhiễm sắc thể trao đổi vật liệu di truyền nhằm chuẩn bị cho quá trình phân chia tế bào . Nếu ADN sai hỏng này không được sửa chữa, tế bào sẽ hoạt động không ổn định rồi có thể dẫn đến cái chết tế bào. Não và hệ thần kinh không thể tái tạo lại số lượng tế bào đã mất đi, do đó phần tiểu não liên quan đến khả năng điều phối chuyển động bị ảnh hưởng.
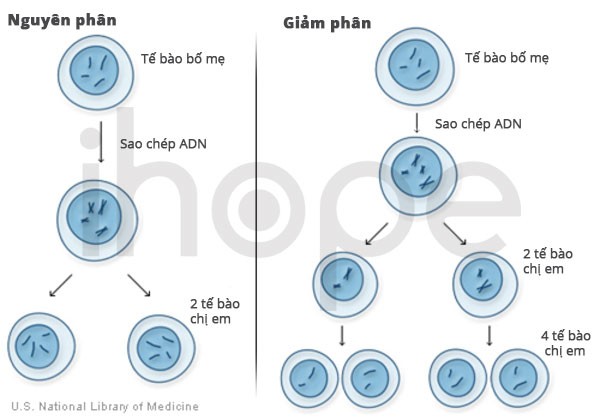
Ảnh: Phân chia tế bào
Nguồn: U.S. National Library of Medicine
Chẩn đoán
Thất điều với mất vận động nhãn cầu được chẩn đoán dựa trên biểu hiện lâm sàng, giai đoạn tiến triển của bệnh kết hợp với tiền sử gia đình. Bên cạnh đó, người bệnh cần thực hiện chẩn đoán phân biệt để tránh nhầm lẫn với các bệnh lý khác như thất điều Friedreich, thất điều do thiếu vitamin E. Ngoài ra, bác sĩ có thể chỉ định bệnh nhân làm xét nghiệm di truyền để xác nhận kết quả chẩn đoán.
Một số phương pháp chẩn đoán bệnh bao gồm:
- Chụp cộng hưởng từ (MRI)
- Chụp X-quang
- Đo nồng độ albumin và cholesterol
Điều trị
Hiện nay, chưa có phương pháp điều trị hoàn toàn bệnh thất điều với mất vận động nhãn cầu. Các liệu pháp chủ yếu nhằm giảm nhẹ triệu chứng và cải thiện chất lượng đời sống bệnh nhân.
Một số phương pháp phổ biến bao gồm:
- Vật lý trị liệu
- Chế độ ăn ít cholesterol
- Điều trị hạ đường huyết
Dạng di truyền
Thất điều với mất vận động nhãn cầu di truyền theo kiểu lặn trên nhiễm sắc thể thường. Do đó, bệnh chỉ biểu hiện khi có cả hai bản sao của gen đột biến trong mỗi tế bào. Bệnh nhân có bố và mẹ mang một bản sao của gen đột biến, nhưng bố mẹ ít khi biểu hiện triệu chứng bệnh.

Nguồn: U.S. National Library of Medicine
Phòng ngừa
Thất điều với mất vận động nhãn cầu di truyền lặn do đột biến gen APTX, SETX hoặc PNKP. Cha mẹ thường mang đột biến dị hợp nên gần như không có biểu hiện bệnh, do đó rất khó phát hiện cho đến khi sinh con. Để chủ động phòng ngừa, cha mẹ nên làm xét nghiệm sàng lọc gen lặn để chủ động cho tương lai của con. Các cặp vợ chồng trước khi mang thai cần tư vấn và xét nghiệm di truyền nhằm đảm bảo sinh con khỏe mạnh. Người thuộc nhóm nguy cơ mắc bệnh cần khám sức khỏe và tầm soát bệnh định kỳ.
Các tên gọi khác
- Adult onset ataxia with oculomotor apraxia
- EAOH
- Early-onset ataxia with ocular motor apraxia and hypoalbuminemia
- SCAN2
- SCAR1
- Spinocerebellar ataxia with axonal neuropathy type 2
- Spinocerebellar ataxia, recessive, non-Friedreich type 1
References
- Genetic Testing Information. Ataxia, early-onset, with oculomotor apraxia and hypoalbuminemia. Retrieved September 14, 2023 from https://www.ncbi.nlm.nih.gov/gtr/conditions/C1859598/
- Genetic and Rare Diseases Information Center. Ataxia-oculomotor apraxia type 1. Retrieved September 14, 2023 from https://rarediseases.info.nih.gov/diseases/9283/ataxia-with-oculomotor-apraxia-type-1/
- Catalog of Genes and Diseases from OMIM. ATAXIA, EARLY-ONSET, WITH OCULOMOTOR APRAXIA AND HYPOALBUMINEMIA; EAOH. Retrieved September 14, 2023 from https://omim.org/entry/208920
- U.S National Library of Medicine. Ataxia with oculomotor apraxia. Retrieved September 14, 2023 from https://medlineplus.gov/genetics/condition/ataxia-with-oculomotor-apraxia/
- Frontiers. Ataxia with oculomotor apraxia type 2 caused by a novel homozygous mutation in SETX gene, and literature review. Retrieved September 14, 2023 from https://www.frontiersin.org/articles/10.3389/fnmol.2022.1019974/full
- MalaCards. Ataxia, Early-Onset, with Oculomotor Apraxia and Hypoalbuminemia (EAOH) Retrieved September 14, 2023 from https://www.malacards.org/card/ataxia_early_onset_with_oculomotor_apraxia_and_hypoalbuminemia
- National Institute of Health. Ataxia with Oculomotor Apraxia Type 1. Retrieved September 14, 2023 from https://www.ncbi.nlm.nih.gov/books/NBK1456/
- National Organization for Rare Disorders. Apraxia. Retrieved September 14, 2023 from https://rarediseases.org/rare-diseases/apraxia/
- Orphanet. Ataxia-oculomotor apraxia type 1. Retrieved September 14, 2023 from https://www.orpha.net/consor/cgi-bin/OC_Exp.php?Lng=GB&Expert=1168





