
Hội chứng Opitz G/BBB (Opitz G/BBB syndrome) là bệnh di truyền hiếm gặp với những biểu hiện đặc trưng như khuôn mặt dị biệt, bất thường liên quan đến hệ hô hấp và hệ tiết niệu-sinh dục, chậm phát triển cũng như sa sút trí tuệ. Hội chứng được phân thành hai thể dựa trên đột biến gây bệnh và dạng di truyền.
Biểu hiện lâm sàng
Phần lớn người mắc hội chứng Opitz G/BBB đều có các biểu hiện lâm sàng tương tự nhau.
Người bệnh thường có những đặc điểm dị biệt trên khuôn mặt như:
- Hai mắt cách xa nhau
- Sứt môi và hở hàm ếch
- Trán dô
- Đường chân tóc hình chữ V
- Sống mũi thấp
- Môi trên mỏng
- Tai thấp
Ngoài ra, hội chứng còn dẫn đến một số biểu hiện khác bao gồm:
- Dị dạng thanh quản, khí quản hoặc thực quản
- Cơ quan sinh dục nam bất thường
- Bệnh lí về đường tiết niệu
- Tắc nghẽn hậu môn
- Dị tật tim
- Dị tật não
- Chậm phát triển, sa sút trí tuệ mức độ nhẹ
- Chậm phát triển vận động
- Rối loạn phổ tự kỷ
Độ phổ biến
Ước tính tỉ lệ nam giới mắc hội chứng Opitz G/BBB thể liên kết nhiễm sắc thể X khoảng 1/10.000–50.000 người. Tuy nhiên, số ca mắc thực tế có thể cao hơn do một số trường hợp bệnh không được chẩn đoán. Tỉ lệ mắc hội chứng Optiz G/BBB thể trội hiện nay chưa được xác định rõ.
Nguyên nhân
Hội chứng Opitz G/BBB thể liên kết nhiễm sắc thể X
Hội chứng thể liên kết nhiễm sắc thể X bắt nguồn từ đột biến trên gen MID1. Gen MID1 cung cấp hướng dẫn tạo ra protein midline-1. Protein này liên kết với các vi ống nhằm kiểm soát quá trình tái sử dụng protein trong cơ thể thay vì phân giải chúng. Vi ống là thành phần của khung xương tế bào, giúp tế bào duy trì hình dạng, hỗ trợ quá trình phân chia và di chuyển của tế bào. Đột biến gen MID1 làm giảm chức năng protein midline-1, do đó cản trở hoạt động tái sử dụng protein, dẫn đến tích tụ protein và ảnh hưởng chức năng của vi ống.
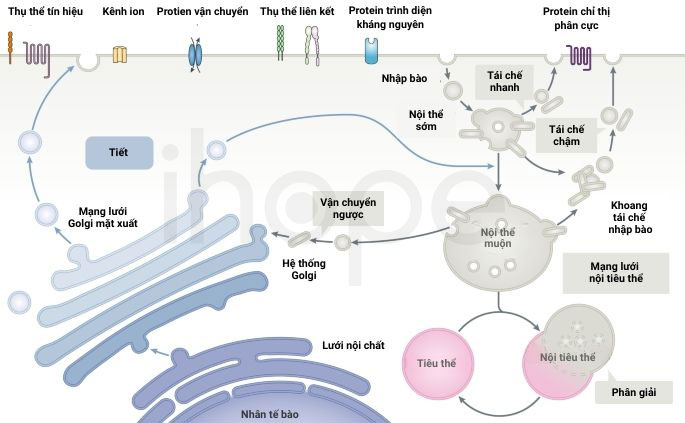
Nguồn: Nature
Hội chứng Opitz G/BBB thể trội
Hội chứng thể trội thường do đột biến mất đoạn trên vùng 11.2 thuộc cánh dài của nhiễm sắc thể số 22 gây ra. Do đó, hội chứng Optiz G/BBB thể trội do đột biến này được phân loại là một dạng của hội chứng mất đoạn 22q11.2. Tuy nhiên, hiện nay người ta chưa xác định được gen nào trong đoạn bị mất dẫn đến các triệu chứng bệnh.
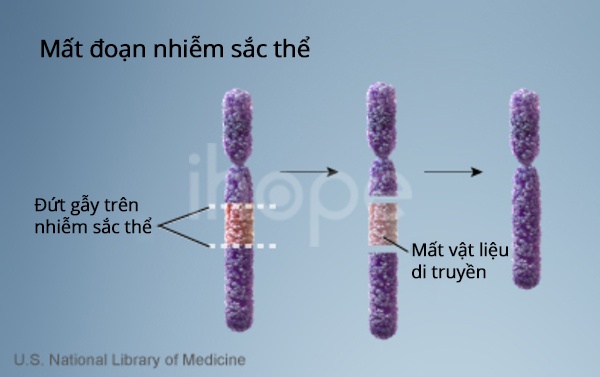
Nguồn: U.S. National Library of Medicine
Mặt khác, người ta ghi nhận hội chứng Opitz G/BBB thể trội có liên quan đến gen SPECC1L gần vùng 22q11.2. Gen này cung cấp hướng dẫn tạo ra cytospin-A—protein tương tác với các thành phần của khung xương tế bào nhằm ổn định vi ống. Khi đột biến gen SPECC1L xuất hiện, cytospin-A giảm tương tác với các thành phần khung xương tế bào. Điều này ảnh hưởng đến cấu trúc sắp xếp thông thường của vi ống và khiến tế bào khó di chuyển đến vị trí cần thiết. Người ta dự đoán đột biến gen SPECC1L có thể là nguyên nhân gây sứt môi và hở hàm ếch đối với một số bệnh nhân mắc hội chứng Opitz G/BBB nhưng chưa thể hiểu rõ đột biến này dẫn đến các đặc điểm khác của hội chứng như thế nào.
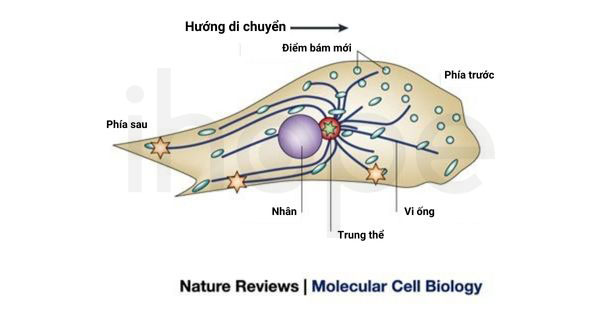
Nguồn: Nature
Ngoài ra, một số người bệnh mắc hội chứng Opitz G/BBB không xuất hiện những dạng đột biến trên. Nguyên nhân gây bệnh trong các trường hợp này vẫn chưa được xác định.
Chẩn đoán
Hội chứng Opitz G/BBB được chẩn đoán dựa trên biểu hiện lâm sàng kết hợp với kết quả chẩn đoán hình ảnh và xét nghiệm di truyền nhằm xác nhận kết quả chẩn đoán.
Chẩn đoán hình ảnh
Kết quả xét nghiệm hình ảnh với những dấu hiệu sau giúp bác sĩ bước đầu chẩn đoán hội chứng Optiz G/BBB:
- Hệ tiết niệu: trào ngược bàng quang niệu quảng, thận ứ nước
- Não: bất sản thể chai, bất sản hoặc giảm sản thuỳ nhộng tiểu não
Xét nghiệm di truyền
Bác sĩ chỉ định xét nghiệm đơn gen nhằm phát hiện đột biến MID1 hoặc xét nghiệm đa gen nhằm xác định các đột biến gây bệnh khác cũng như loại trừ khả năng mắc một số hội chứng tương tự.
Điều trị
Hiện nay, chưa có phương pháp điều trị hoàn toàn hội chứng Opitz G/BBB. Các liệu pháp chủ yếu nhằm giảm nhẹ triệu chứng và cải thiện chất lượng đời sống người bệnh. Trẻ bị sứt môi hoặc hở hàm ếch cần được kiểm tra thính lực định kì nhằm phát hiện và can thiệp sớm các vấn đề về khả năng nghe.
Ngoài ra, những biện pháp sau có thể hỗ trợ người bệnh cải thiện một số triệu chứng:
- Phẫu thuật chỉnh hình các dị tật khuôn mặt, hệ tiết niệu-sinh dục
- Phẫu thuật mở khí quản
- Dùng thuốc điều trị trào ngược dạ dày thực quản
- Trị liệu ngôn ngữ
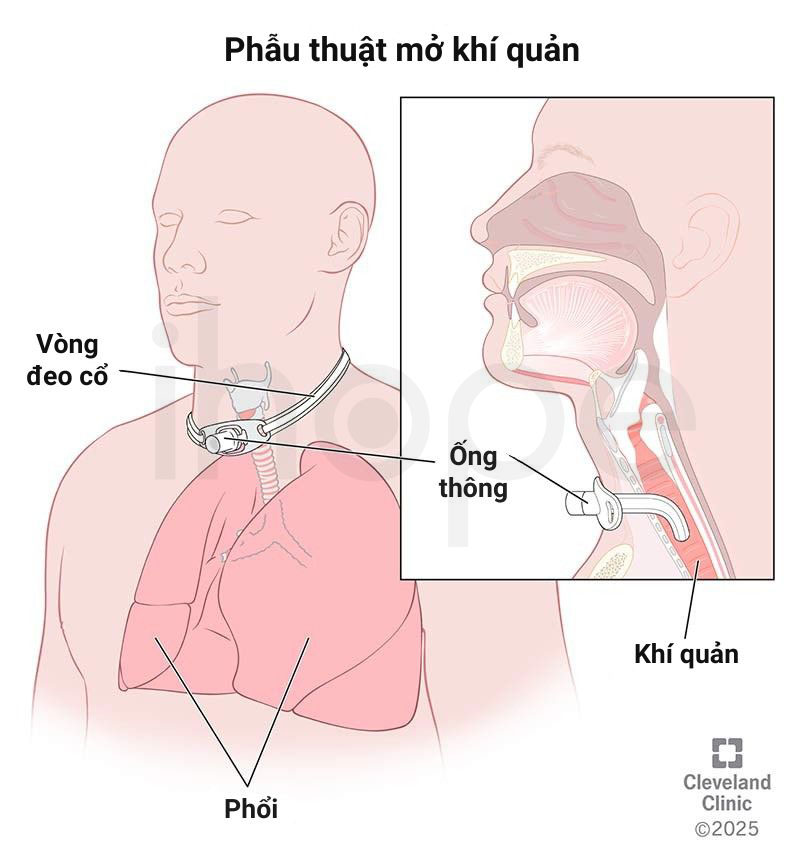
Nguồn: Cleveland Clinic
Dạng di truyền
Hội chứng Opitz G/BBB di truyền theo kiểu lặn liên kết với nhiễm sắc thể giới tính X. Nam giới chỉ có một nhiễm sắc thể X, do đó một bản sao của gen MID1 đột biến trong mỗi tế bào đủ gây ra triệu chứng nghiêm trọng bệnh. Người cha bị bệnh không thể truyền gen đột biến này cho con trai. Đối với phụ nữ có hai nhiễm sắc thể X, đột biến tại một bản sao của gen MID1 thường chỉ gây ra các triệu chứng nhẹ.
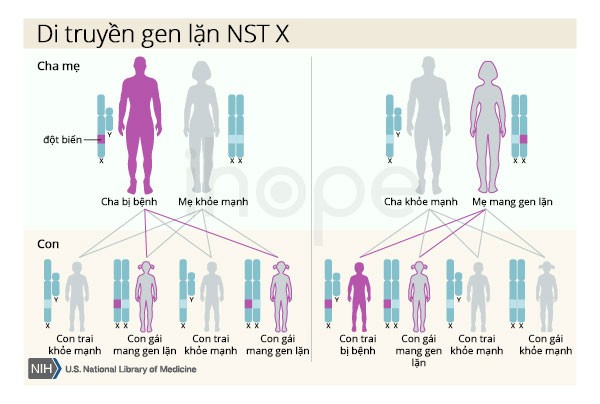
Nguồn: U.S. National Library of Medicine
Ngoài ra, một số ít trường hợp hội chứng Opitz G/BBB di truyền theo kiểu trội trên nhiễm sắc thể thường. Do đó, một bản sao của gen đột biến trong mỗi tế bào đủ để gây ra bệnh. Người bệnh thường được di truyền từ cha hoặc mẹ mắc bệnh. Một số ít trường hợp là do đột biến mới (de novo) xuất hiện trong quá trình hình thành tế bào sinh sản (trứng, tinh trùng) hoặc quá trình phát triển phôi sớm.

Nguồn: U.S. National Library of Medicine
Phòng ngừa
Hội chứng Opitz G/BBB có cơ chế di truyền liên kết X phức tạp nên khó phát hiện trên những người phụ nữ mang gen bệnh cho đến khi sinh con. Để chủ động phòng ngừa, người mẹ nên làm xét nghiệm sàng lọc gen lặn cho tương lai của con. Các cặp vợ chồng trước khi mang thai cần tư vấn và xét nghiệm di truyền đảm bảo sinh con khỏe mạnh. Người thuộc nhóm nguy cơ mắc bệnh cần khám sức khỏe và tầm soát bệnh định kì.
Các tên gọi khác
- Hypertelorism with esophageal abnormalities and hypospadias
- Hypertelorism-hypospadias syndrome
- Hypospadias-dysphagia syndrome
- Opitz BBB syndrome
- Opitz BBB/G syndrome
- Opitz G syndrome
- Opitz syndrome
- Opitz-Frias syndrome
References
- National Library of Medicine. MID1-Related Opitz G/BBB Syndrome. Retrieved August 15, 2025 from https://www.ncbi.nlm.nih.gov/books/NBK1327/
- National Organization for Rare Disorders. X-linked Opitz G/BBB Syndrome. Retrieved August 15, 2025 from https://rarediseases.org/rare-diseases/x-linked-opitz-gbbb-syndrome/
- OMIM. OPITZ GBBB SYNDROME; GBBB . Retrieved August 15, 2025 from https://omim.org/entry/300000
- U.S National Library of Medicine. Opitz G/BBB syndrome. Retrieved August 15, 2025 from https://medlineplus.gov/genetics/condition/opitz-g-bbb-syndrome
- Orphanet. Opitz GBBB syndrome. Retrieved August 15, 2025 from https://www.orpha.net/en/disease/detail/2745





