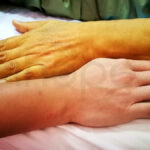
Axit mật kích thích bài tiết, vận chuyển dịch mật, hỗ trợ hấp thụ chất béo và vitamin tan trong chúng (vitamin A, D, E, K). Khiếm khuyết tổng hợp axit mật bẩm sinh loại 1 (congenital bile acid synthesis defect type 1) làm suy yếu quá trình sản xuất và giải phóng dịch mật từ tế bào gan. Triệu chứng đặc trưng của bệnh là gan ứ mật.
Nguồn: U.S. National Library of Medicine
Biểu hiện lâm sàng
Các dấu hiệu của bệnh khởi phát trong tuần đầu sau sinh hoặc tại thời điểm bất kỳ từ giai đoạn trẻ nhỏ đến tuổi trưởng thành. Trẻ sơ sinh biểu hiệu triệu chứng chậm phát triển, vàng da do lưu lượng mật suy giảm và hình thành tích tụ mật một phần hoặc phân chứa chất béo. Khi bệnh tiến triển, người bệnh có thể phát triển các bất thường về gan (gan to, viêm gan), lách to, còi xương .
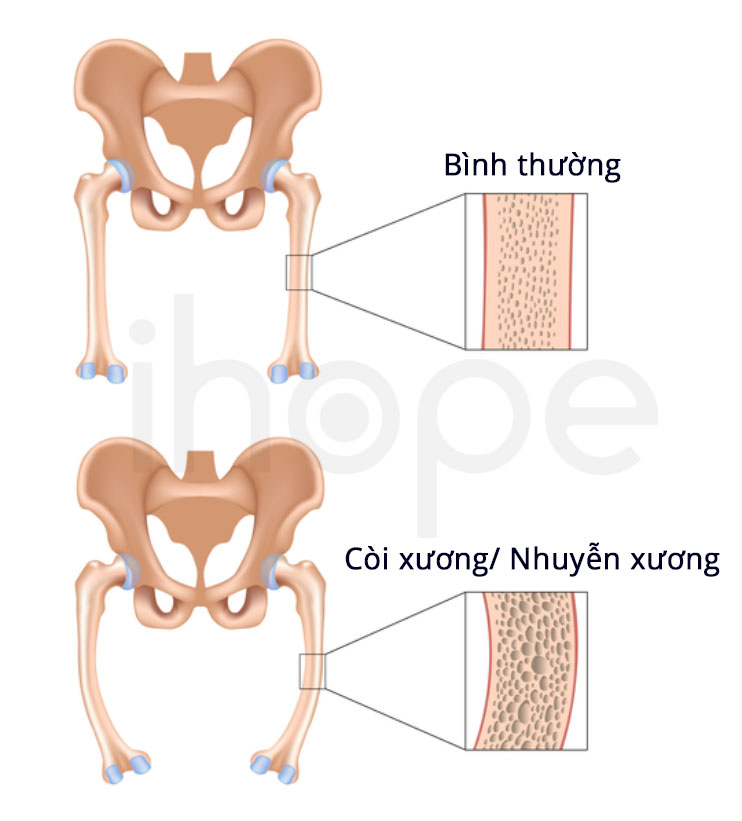
Bệnh còi xương
Nguồn: Alila Medical Media/Shutterstock.com
Trẻ mắc bệnh không được điều trị kịp thời có thể dẫn đến xơ gan hoặc tử vong.
Độ phổ biến
Tỷ lệ khiếm khuyết tổng hợp axit mật bẩm sinh loại 1 chưa được thống kê cụ thể, tuy nhiên nó là dạng phổ biến nhất trong tất cả các bệnh tổng hợp axit mật bẩm sinh. Do đó, tỉ lệ mắc bệnh chung từ 1 đến 9 trên một triệu người.
Nguyên nhân
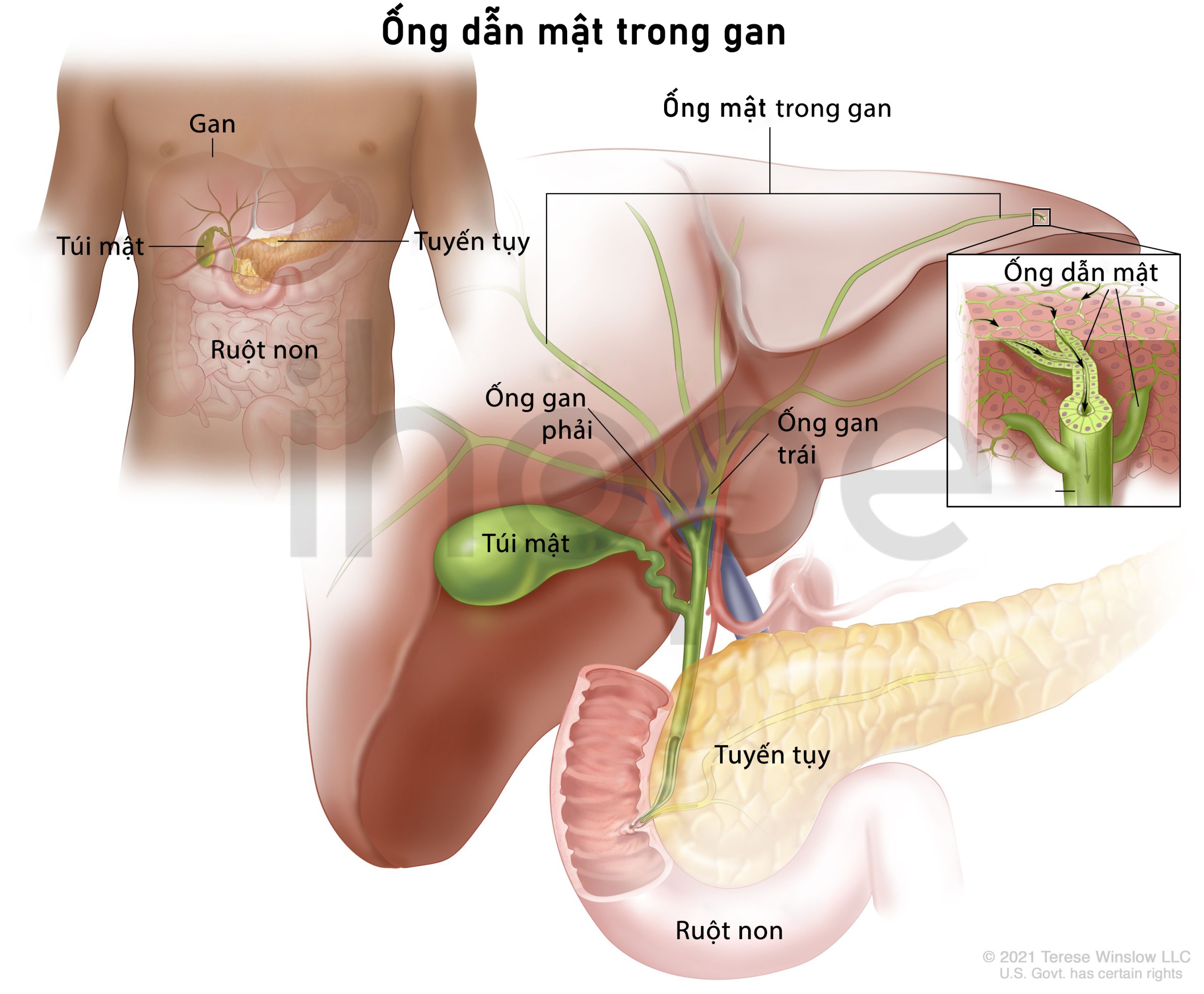
Đột biến gen HSD3B7 gây ra khiếm khuyết tổng hợp axit mật bẩm sinh loại 1. Gen HSD3B7 cung cấp hướng dẫn tạo ra enzyme 3β-HSD7 (3 beta-hydroxysteroid dehydrogenase type 7) trong các tế bào gan tham gia sản xuất axit mật. Cholesterol được chuyển hoá thành axit mật thông qua quá trình gồm nhiều bước. Tại bước thứ hai, enzyme 3β-HSD7 xử lí 7alpha(α)-hydroxycholesterol thành 7α-hydroxy-4-cholesten-3-one.
Đột biến gen HSD3B7 dẫn đến giảm sản enzyme 3β-HSD7 hoặc tạo ra phiên bản enzyme bất hoạt. Thiếu enzyme 3β-HSD7 khiến quá trình xử lý cholesterol suy yếu. Do đó, 7α-hydroxycholesterol được chuyển hoá thành các axit mật bất thường nên chúng không thể đi ra khỏi gan để vào ruột—nơi cần axit mật để phân giải chất béo. Những vitamin tan trong chất béo không được hấp thụ góp phần gây ra các dấu hiệu và triệu chứng của bệnh.
Chẩn đoán
Các trường hợp sau nên được chẩn đoán rối loạn tổng hợp axit mật:
- Trẻ sơ sinh hoặc trẻ nhỏ bị vàng da
- Các triệu chứng khác của gan ứ mật không rõ nguyên nhân
- Thiếu vitamin tan trong chất béo
- Chậm phát triển
Bác sĩ có thể dựa vào các triệu chứng đặc trưng, bệnh sử chi tiết của bệnh nhân và đánh giá lâm sàng để đưa ra chẩn đoán sau cùng. Tuy nhiên, các triệu chứng của khiếm khuyết tổng hợp axit mật bẩm sinh loại 1 trùng lặp với nhiều bệnh gan khác. Do đó, kết quả chẩn đoán nên được xác nhận bằng xét nghiệm tại các phòng thí nghiệm chẩn đoán chuyên sâu.
Phát hiện sớm và chẩn đoán kịp thời là cực kỳ quan trọng, bởi vì nhiều trường hợp cho thấy đáp ứng mạnh với liệu pháp thay thế axit mật qua đường uống.
Điều trị
Hiện nay chưa có phương pháp điều trị hoàn toàn khiếm khuyết tổng hợp axit mật bẩm sinh loại 1. Do đó, mục tiêu điều trị tập trung làm giảm triệu chứng, giúp cải thiện chất lượng cuộc sống của người bệnh. Chế độ ăn uống hạn chế chất béo và cholesterol, tăng cường lượng protein, bổ sung axit mật tổng hợp và các dẫn xuất của nó. Người bệnh có thể bổ sung axit mật tổng hợp như axit cholic theo hướng dẫn của bác sĩ. Bên cạnh đó, người bệnh nên thăm khám sức khỏe định kỳ nhằm đánh giá hiệu quả điều trị và phòng ngừa các biến chứng tiềm ẩn.
Dạng di truyền
Khiếm khuyết tổng hợp axit mật bẩm sinh loại 1 di truyền theo kiểu lặn trên nhiễm sắc thể thường. Do đó, bệnh chỉ biểu hiện khi có cả hai bản sao của gen đột biến trong mỗi tế bào. Bệnh nhân mắc bệnh lặn trên nhiễm sắc thể thường sẽ có bố và mẹ mang một bản sao của gen đột biến, nhưng bố mẹ ít khi biểu hiện triệu chứng bệnh.

Nguồn: U.S. National Library of Medicine
Phòng ngừa
Bệnh di truyền lặn do đột biến gen HSD3B7, cha mẹ mang đột biến dị hợp nên gần như không có biểu hiện bệnh, do đó rất khó phát hiện cho đến khi sinh con. Để chủ động phòng ngừa, cha mẹ nên làm xét nghiệm sàng lọc gen lặn để chủ động cho tương lai của con. Các cặp vợ chồng trước khi mang thai cần tư vấn và xét nghiệm di truyền đảm bảo sinh con khỏe mạnh. Người thuộc nhóm nguy cơ mắc bệnh cần khám sức khỏe và tầm soát bệnh định kỳ.
Các tên gọi khác
- 3beta-HSDH deficiency
- 3beta-hydroxy-delta-5-C27-steroid dehydrogenase deficiency
- 3beta-hydroxy-delta-5-C27-steroid oxidoreductase deficiency
- CBAS1
References
- Genetic Testing Information. Congenital bile acid synthesis defect 1. Retrieved 17 May 2023 from https://www.ncbi.nlm.nih.gov/gtr/conditions/C1843116/
- Genetic and Rare Diseases Information Center. Congenital bile acid synthesis defect, type 1. Retrieved 17 May 2023 from https://rarediseases.info.nih.gov/diseases/9813/congenital-bile-acid-synthesis-defect-type-1
- Catalog of Genes and Diseases from OMIM. BILE ACID SYNTHESIS DEFECT, CONGENITAL, 1. Retrieved 17 May 2023 from https://omim.org/entry/607765
- U.S National Library of Medicine. Congenital bile acid synthesis defect type 1. Retrieved 17 May 2023 from https://medlineplus.gov/genetics/condition/congenital-bile-acid-synthesis-defect-type-1/
- MalaCards. Bile Acid Synthesis Defect, Congenital, 1 (CBAS1). Retrieved 17 May 2023 from https://www.malacards.org/card/bile_acid_synthesis_defect_congenital_1
- National Organization for Rare Disorders. Bile Acid Synthesis Disorders. Retrieved 17 May 2023 from https://rarediseases.org/rare-diseases/bile-acid-synthesis-disorders/
- Orphanet. Congenital bile acid synthesis defect type 1. Retrieved 17 May 2023 from https://www.orpha.net/consor/cgi-bin/OC_Exp.php?lng=en&Expert=79301
- Cheng JB, Jacquemin E, Gerhardt M, Nazer H, Cresteil D, Heubi JE, Setchell KD, Russell DW. Molecular genetics of 3beta-hydroxy-Delta5-C27-steroid oxidoreductase deficiency in 16 patients with loss of bile acid synthesis and liver disease. J Clin Endocrinol Metab. 2003 Apr;88(4):1833-41. doi: 10.1210/jc.2002-021580
- Clayton PT. Disorders of bile acid synthesis. J Inherit Metab Dis. 2011 Jun;34(3):593-604. doi: 10.1007/s10545-010-9259-3
- Subramaniam P, Clayton PT, Portmann BC, Mieli-Vergani G, Hadzic N. Variable clinical spectrum of the most common inborn error of bile acid metabolism--3beta-hydroxy-Delta 5-C27-steroid dehydrogenase deficiency. J Pediatr Gastroenterol Nutr. 2010 Jan;50(1):61-6. doi: 10.1097/MPG.0b013e3181b47b34