Loạn dưỡng cơ Miyoshi (Miyoshi myopathy) là bệnh di truyền làm suy yếu các cơ nằm xa trung tâm cơ thể, chủ yếu là cơ chân. Bệnh thường khởi phát trong độ tuổi trưởng thành.
Biểu hiện lâm sàng
Dấu hiệu ban đầu của loạn dưỡng cơ Miyoshi gồm yếu và teo cơ trên một hoặc hai bắp chân. Trong trường hợp teo cơ một bên chân, hai chân người bệnh có kích thước bắp chân không đều. Yếu cơ bắp chân khiến bệnh nhân không thể nhón chân.
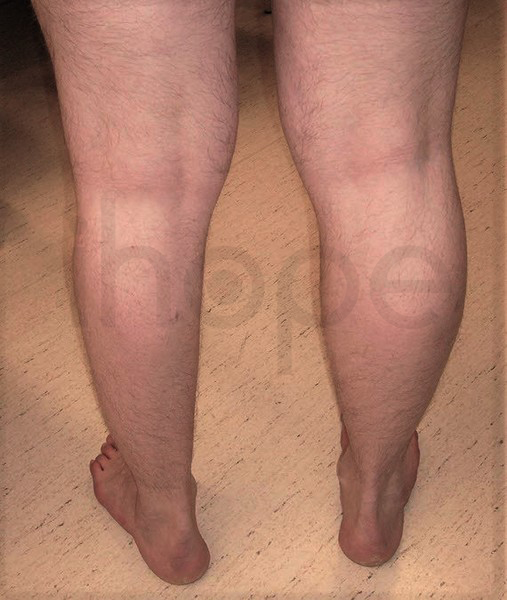
có kích thước không đều
Nguồn: GeneReviews, © 1993-2020 University of Washington
Khi bệnh tiến triển chậm, triệu chứng yếu, teo cơ lan dần lên đùi và mông. Bệnh có thể tác động đến nhóm cơ bắp tay và vai. Bệnh nhân có thể gặp khó khăn khi leo cầu thang hoặc đi bộ trong thời gian dài. Một số người bệnh có thể cần sử dụng xe lăn để hỗ trợ di chuyển.
Trong một số trường hợp, người bệnh có biểu hiện loạn nhịp tim. Ngoài ra, người bệnh thường có nồng độ enzyme creatine kinase (CK) trong máu tăng cao. Chúng là dấu hiệu phổ biến cho thấy cơ thể đang tổn thương cơ.
Độ phổ biến
Hiện nay, chưa có thống kê cụ thể về tỉ lệ mắc bệnh loạn dưỡng cơ Miyoshi. Tại Nhật Bản—nơi bệnh được phát hiện, tỉ lệ mắc bệnh khoảng 1/440.000 người.
Nguyên nhân
Đột biến gen DYSF và ANO5 gây ra loạn dưỡng cơ Miyoshi. Nếu do đột biến gen ANO5 gây ra, bệnh được phân loại là loạn dưỡng cơ anoctamin xa. Trong trường hợp do đột biến gen DYSF, bệnh được phân loại là loạn dưỡng cơ dysferlin. Hai gen này cung cấp hướng dẫn tạo ra các protein hiện diện chủ yếu trong cơ xương.
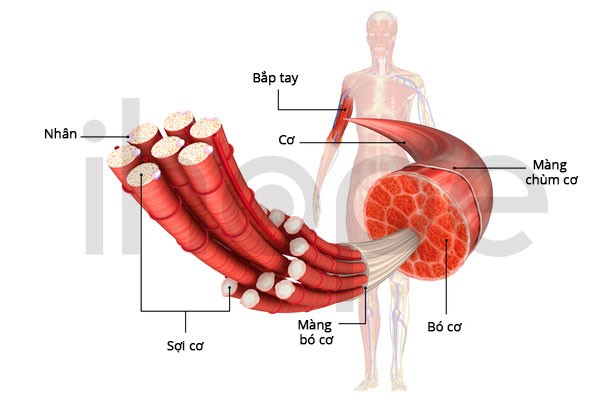
Nguồn: U.S. National Library of Medicine
Gen DYSF
Gen DYSF cung cấp hướng dẫn tạo ra protein dysferlin hiện diện một protein được tìm thấy trong lớp màng sinh chất mỏng bao quanh sợi cơ. Dysferlin có chức năng hỗ trợ sửa chữa màng sinh chất khi lớp màng này hư hỏng hoặc rách do căng cơ.
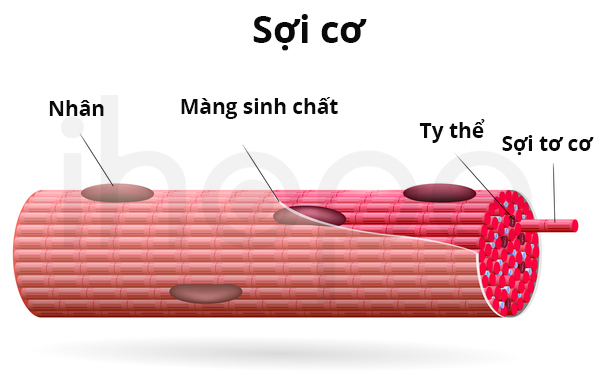
Nguồn: Medlineplus.gov
Gen ANO5
Gen ANO5 cung cấp hướng dẫn tạo ra protein anoctamin-5 nằm trong màng của lưới nội chất. Anoctamin-5 liên quan đến quá trình sản xuất, xử lí và vận chuyển protein. Anoctamin-5 hoạt động như một kênh vận chuyển, chúng cho phép các nguyên tử clo tích điện đi vào và ra khỏi lưới nội chất, qua đó điều hòa dòng chảy clorua trong tế bào cơ. Quá trình điều hòa này có tác dụng kiểm soát hoạt động co thắt và thư giãn cơ.
Nguồn: U.S. National Library of Medicine
Đột biến gen DYSF và ANO5 dẫn đến giảm số lượng protein tạo thành tương ứng hoặc hoàn toàn không có protein. Tình trạng thiếu dysferlin làm giảm khả năng sửa chữa tổn thương màng sarcolemma bao quanh sợi cơ. Do đó, tổn thương tích tụ đến mức gây teo sợi cơ. Người ta chưa rõ tại sao tổn thương màng sarcolemma dẫn đến biểu hiện suy yếu và teo cơ đặc trưng của loạn dưỡng cơ Miyoshi.
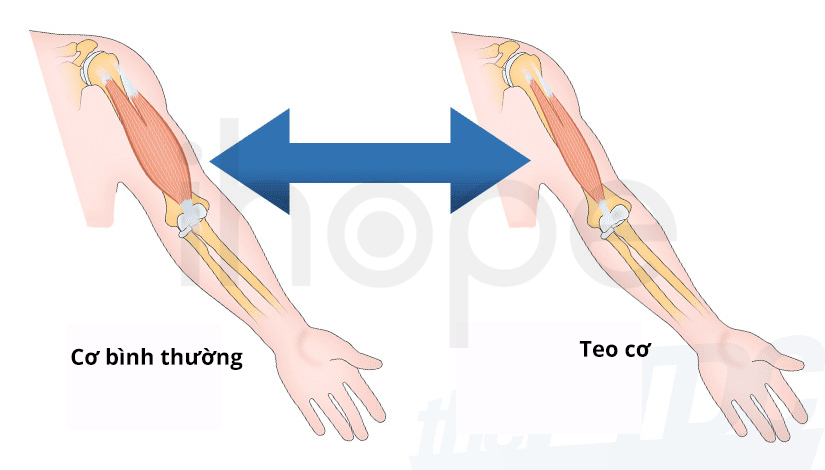
Nguồn: Mount Sinai
Hơn nữa, tác động của mất anoctamin-5 lên cơ thể cũng chưa được hiểu rõ. Tuy clorua cần thiết cho chức năng cơ, người ta vẫn chưa hiểu vì sao thiếu anoctamin-5 gây ra các dấu hiệu và triệu chứng của loạn dưỡng cơ Miyoshi.
Chẩn đoán
Loạn dưỡng cơ Miyoshi được chẩn đoán dựa trên nhiều xét nghiệm và kĩ thuật chuyên sâu. Trong đó, xét nghiệm máu được sử dụng phổ biến nhất trong chẩn đoán. Người bệnh thường có nồng độ enzyme Creatine Kinase (CK) tăng cao gấp 20–150 lần so với người khỏe mạnh. Nồng độ enzyme bất thường là dấu hiệu cho thấy tổn thương cơ đang diễn ra trong cơ thể.
Ngoài ra, bác sĩ cũng tiến hành sinh thiết cơ cho người bệnh. Một mẫu mô cơ nhỏ của bệnh nhân được tách ra và đem phân tích dưới kính hiển vi nhằm tìm kiếm những biến đổi đặc trưng của loạn dưỡng cơ. Mặt khác, nhuộm hóa mô miễn dịch cũng là phương pháp chẩn đoán hữu ích nhằm xác định lượng protein trong mô cơ.
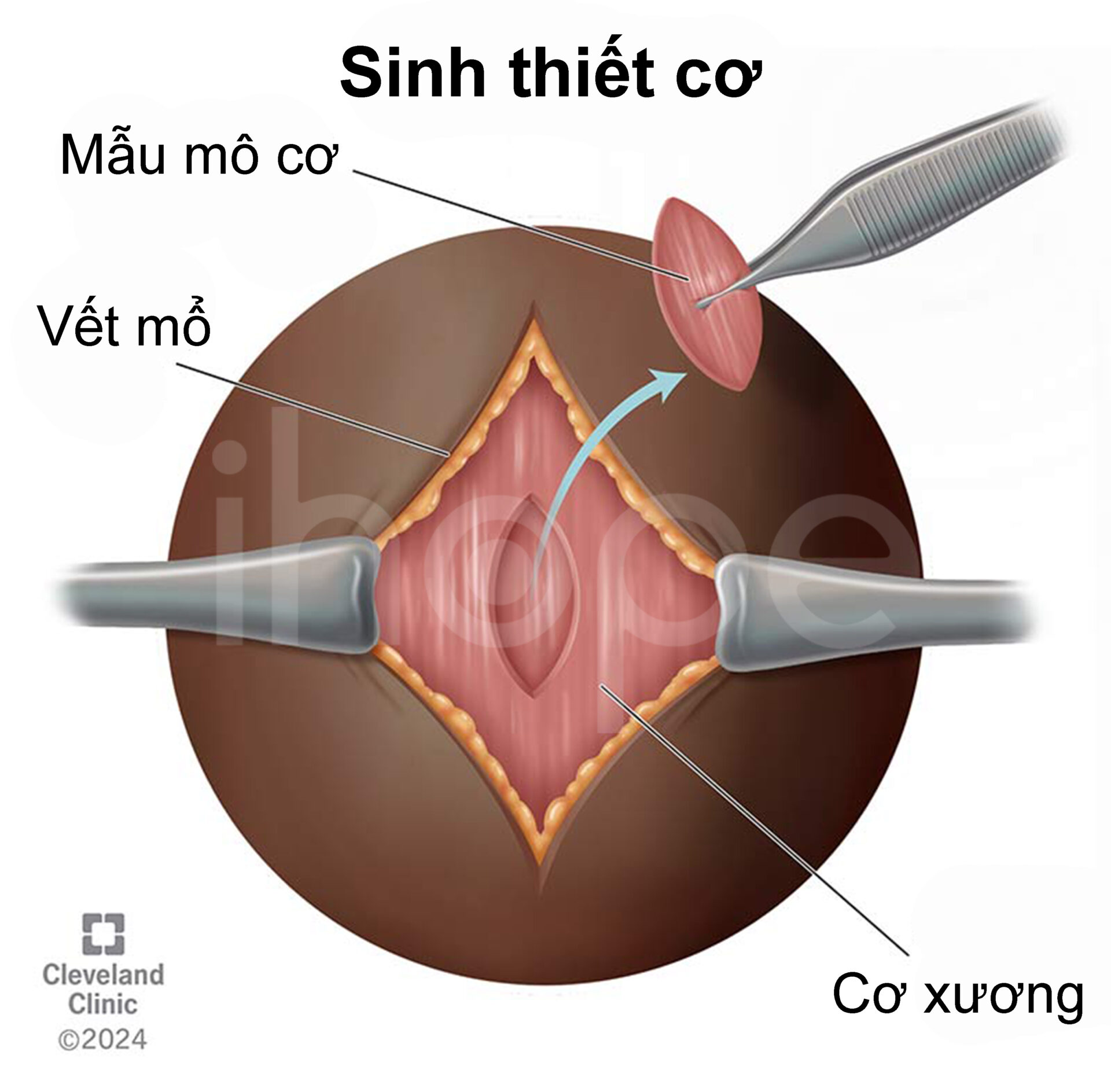
Nguồn: Cleveland Clinic
Trong trường hợp kết quả hóa mô miễn dịch không rõ ràng, kĩ thuật Western blot được sử dụng nhằm xác định mức độ hiện diện và số lượng protein dysferlin hoặc anoctamin-5 trong mô cơ. Khi người bệnh thực hiện chụp cộng hưởng từ (MRI), mô cơ trong bắp chân thường bị thay thế bằng mô mỡ. Điện cơ đồ (EMG) cũng là xét nghiệm phổ biến được sử dụng để phát hiện bất thường trong hoạt động điện cơ.
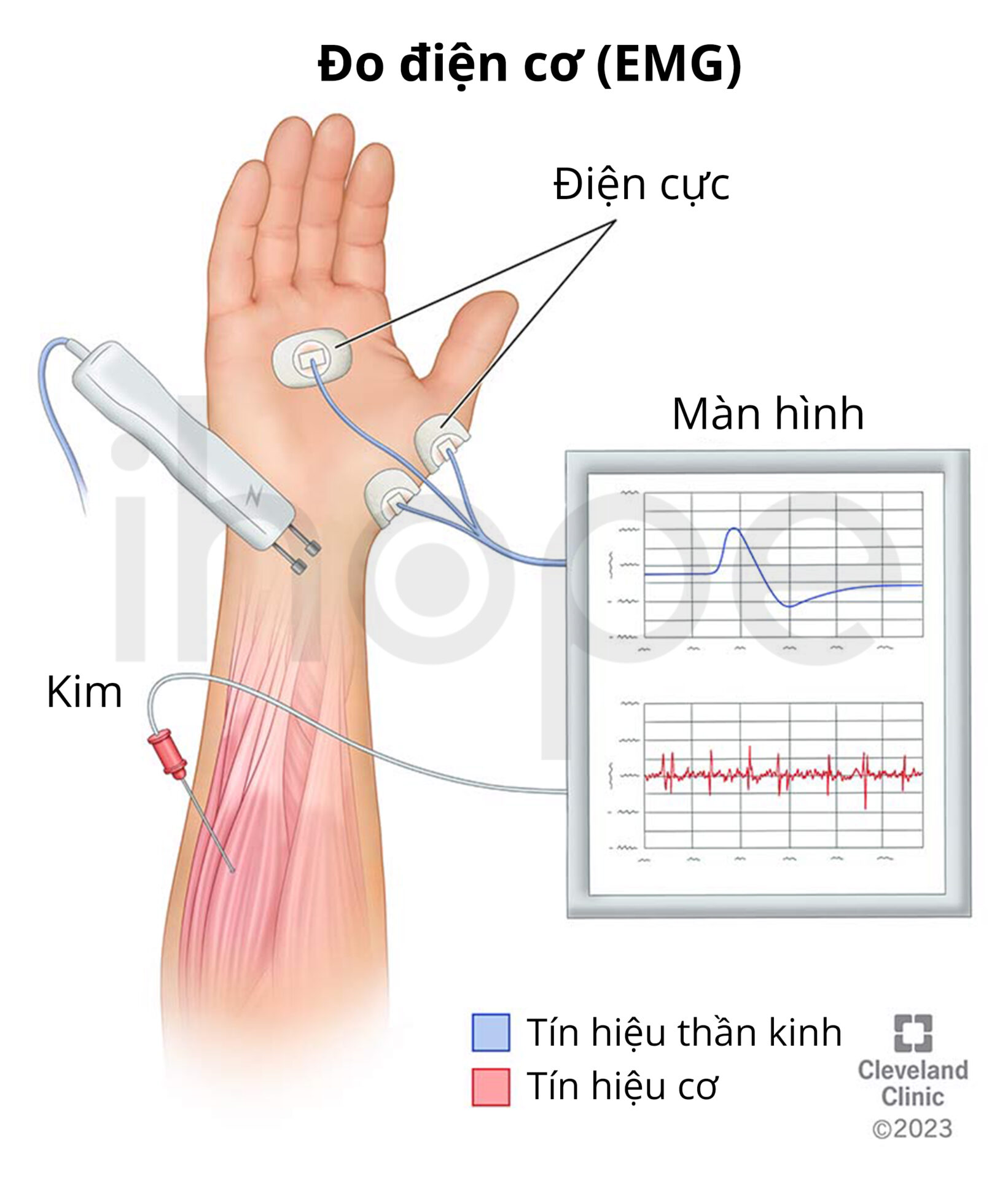
Nguồn: Cleveland Clinic
Ngoài ra, người bệnh có thể thực hiện các xét nghiệm di truyền nhằm phát hiện đột biến gen DYSF hoặc ANO5, qua đó kết quả chẩn đoán được xác nhận.
Điều trị
Hiện nay, chưa có phương pháp điều trị hoàn toàn loạn dưỡng cơ Miyoshi. Các liệu pháp chủ yếu nhằm giảm nhẹ triệu chứng và cải thiện chất lượng đời sống cho người bệnh.
Kiểm soát cân nặng là yếu tố quan trọng trong điều trị và phòng ngừa bệnh loạn dưỡng cơ Miyoshi. Duy trì cân nặng hợp lí và tránh béo phì có thể giảm thiểu áp lực lên hệ cơ của người bệnh. Đồng người, người bệnh cần tiến hành vật lí trị liệu nhằm duy trì khả năng vận động và độ linh hoạt của khớp. Phương pháp này cũng mang lại hiệu quả trong phòng ngừa co cứng cơ tiến triển. Khi bệnh tiến triển nặng, bệnh nhân cần sử dụng các thiết bị hỗ trợ di chuyển như nạng, khung tập đi hoặc xe lăn nếu cần thiết.
Dạng di truyền
Loạn dưỡng cơ Miyoshi di truyền theo kiểu lặn trên nhiễm sắc thể thường. Do đó, bệnh chỉ biểu hiện khi có cả hai bản sao của gen đột biến trong mỗi tế bào. Bệnh nhân mắc bệnh di truyền gen lặn trên nhiễm sắc thể thường có bố và mẹ mang một bản sao của gen đột biến, nhưng họ ít khi biểu hiện triệu chứng bệnh.

Nguồn: U.S. National Library of Medicine
Phòng ngừa
Loạn dưỡng cơ Miyoshi di truyền lặn đột biến gen DYSF hoặc ANO5. Cha mẹ mang đột biến dị hợp nên gần như không có biểu hiện bệnh, do đó rất khó phát hiện cho đến khi sinh con. Cha mẹ nên làm xét nghiệm sàng lọc gen lặn để chủ động cho tương lai của con. Các cặp vợ chồng trước khi mang thai cần tư vấn và xét nghiệm di truyền nhằm đảm bảo sinh con khỏe mạnh. Người thuộc nhóm nguy cơ mắc bệnh cần khám sức khỏe và tầm soát bệnh định kì.
Các tên gọi khác
- MMD
- Miyoshi distal myopathy
- Miyoshi muscular dystrophy
- Distal muscular dystrophy, Miyoshi type
References
- Genetic Testing Information. Miyoshi muscular dystrophy 1. Retrieved February 24, 2025 from https://www.ncbi.nlm.nih.gov/gtr/conditions/C4551973/
- Genetic and Rare Diseases Information Center. Miyoshi myopathy. Retrieved February 24, 2025 from https://rarediseases.info.nih.gov/diseases/9676/index
- Catalog of Genes and Diseases from OMIM. MIYOSHI MUSCULAR DYSTROPHY 1; MMD1. Retrieved February 24, 2025 from https://omim.org/entry/254130
- U.S National Library of Medicine. Miyoshi myopathy. Retrieved February 24, 2025 from https://medlineplus.gov/genetics/condition/miyoshi-myopathy/
- MalaCards. Miyoshi Muscular Dystrophy. Retrieved February 24, 2025 from https://www.malacards.org/card/miyoshi_muscular_dystrophy
- National Institute of Health. Clinical Presentation, Diagnosis, and Genetic Insights of Miyoshi Myopathy: A Case Report and Literature Review. Retrieved February 24, 2025 from https://pmc.ncbi.nlm.nih.gov/articles/PMC11457810/
- National Institute of Health. Miyoshi distal muscular dystrophy (Miyoshi myopathy). Retrieved February 24, 2025 from https://pubmed.ncbi.nlm.nih.gov/21301039/
- Orphanet. Miyoshi myopathy. Retrieved February 24, 2025 from https://www.orpha.net/en/disease/detail/45448
- ResearchGate. Miyoshi myopathy: diagnosis of a familial case of dysferlinopathy. Retrieved February 24, 2025 from https://www.researchgate.net/publication/301645768