Rối loạn dự trữ axit sialic tự do (Free sialic acid storage disorder) là bệnh di truyền chủ yếu ảnh hưởng đến não và tủy sống (hệ thần kinh trung ương). Bệnh này thuộc nhóm bệnh rối loạn dự trữ lysosome. Mỗi dạng bệnh trong nhóm này đều do thiếu một enzyme hoặc protein lysosome. Biểu hiện lâm sàng thay đổi tùy theo từng bệnh nhân; bệnh được phân thành ba thể dựa trên mức độ nghiêm trọng.
Biểu hiện lâm sàng
Các triệu chứng của bệnh rối loạn dự trữ axit sialic tự do rất đa dạng. Dạng nghiêm trọng nhất mang tên infantile free sialic acid storage disease hay ISSD. Các triệu chứng thường khởi phát trong giai đoạn sơ sinh. Một số trường hợp xuất hiện phù thai (hiện tượng tích tụ dịch dư thừa trong cơ thể) trước hoặc ngay sau khi sinh. Trẻ sơ sinh mắc dạng này thường chậm phát triển nghiêm trọng, giảm trương lực cơ và tăng trưởng kém. Các biểu hiện khác bao gồm khuôn mặt thô, dị dạng xương, gan lách to, tim to và co giật. Nhiễm trùng đường hô hấp xảy ra thường xuyên và có thể đe dọa tính mạng. Trẻ mắc thể bệnh này thường chỉ sống sót đến giai đoạn trẻ nhỏ.
Trẻ mắc dạng nhẹ nhất, còn gọi là bệnh Salla, có thể không biểu hiện triệu chứng khi mới sinh. Mặc dù thời điểm khởi phát bệnh có thể khác nhau giữa các cá nhân, nhưng giảm trương lực cơ xảy ra phổ biến trong năm đầu đời. Bệnh nhân mắc thể này có khuyết tật trí tuệ và chậm phát triển, từ đó gây khó khăn trong quá trình tập đi và tập nói. Các dấu hiệu khác bao gồm vấn đề về thất điều, căng cơ bất thường và cử động tứ chi không tự chủ (múa vờn). Các triệu chứng thường tiến triển nặng dần theo thời gian. Mặc dù tuổi thọ có thể bị rút ngắn, người mắc bệnh Salla thường sống sót đến tuổi trưởng thành.
Người mắc dạng trung gian, hay còn gọi là bệnh Salla thể nặng trung gian, có các dấu hiệu và triệu chứng nghiêm trọng hơn bệnh Salla nhưng nhẹ hơn so với ISSD.
Độ phổ biến
Hiện nay người ta ghi nhận ít hơn 300 trường hợp mắc bệnh rối loạn dự trữ axit sialic tự do. Bệnh Salla phổ biến hơn trong nhóm người gốc Phần Lan và Thụy Điển với khoảng 150 ca bệnh được báo cáo.
Nguyên nhân
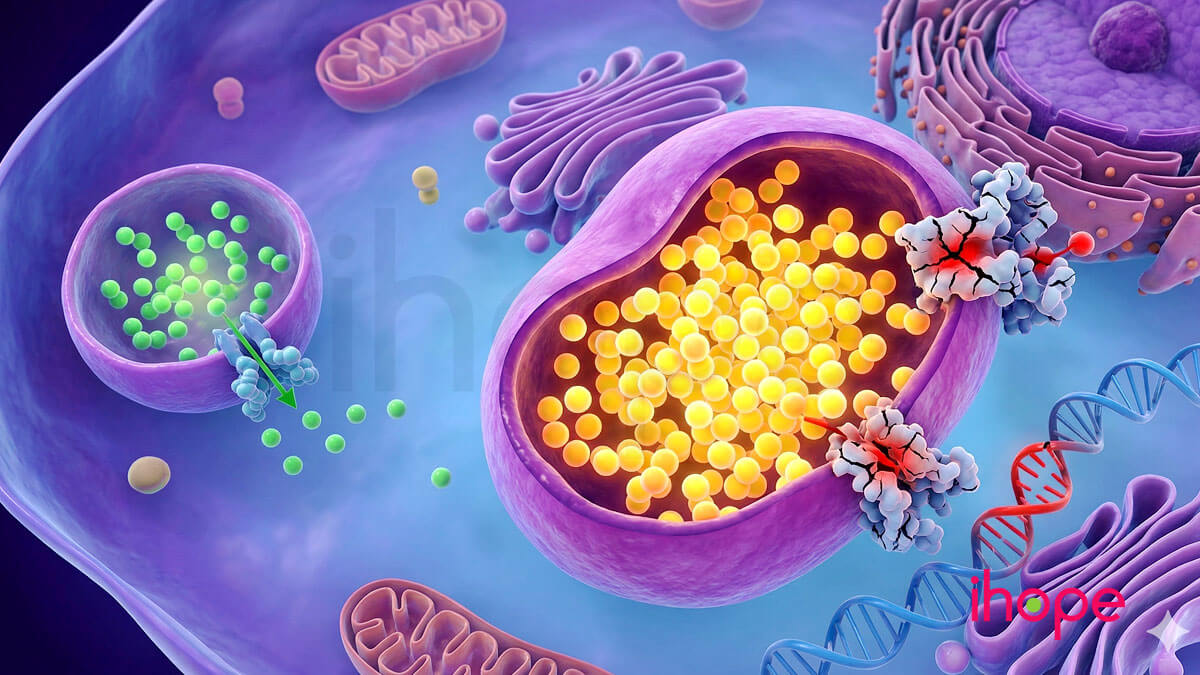
Đột biến gen SLC17A5 gây ra tất cả các dạng của bệnh rối loạn dự trữ axit sialic tự do. Gen SLC17A5 cung cấp hướng dẫn tạo ra protein sialin. Protein này nằm trên màng lysosome - các khoang trong tế bào chịu trách nhiệm tiêu hóa và tái chế các phân tử khác nhau.
Sialin vận chuyển axit sialic tự do đến các bộ phận khác của tế bào. Axit sialic được tạo ra khi chất béo và protein bị phân giải, đóng vai trò quan trọng trong giao tiếp và sự kết dính giữa các tế bào. Axit sialic được gọi là tự do khi không gắn kết với các phân tử khác.
Một số đột biến gen SLC17A5 khiến tế bào sản xuất protein sialin mất chức năng, trong khi các đột biến khác làm gián đoạn quá trình sản xuất sialin. Trong một vài trường hợp, sialin được tạo ra nhưng không thể di chuyển đến màng lysosome.
Các biến thể gen SLC17A5 làm thay đổi hoặc mất hoạt tính của sialin khiến axit sialic tự do tích tụ trong lysosome. Người ta chưa rõ vì sao sự tích tụ này gây ra các dấu hiệu thần kinh của bệnh.
Chẩn đoán
Bác sĩ có thể nghi ngờ bệnh rối loạn dự trữ axit sialic tự do dựa trên các dấu hiệu lâm sàng đặc trưng, tiền sử bệnh cùng kết quả đánh giá sức khỏe toàn diện. Ngoài ra, các xét nghiệm chuyên biệt giúp phát hiện nồng độ axit sialic tự do tăng cao trong tế bào, mô hoặc nước tiểu cũng đóng vai trò quan trọng giúp nhận diện bệnh.
Chẩn đoán trước sinh có thể thực hiện thông qua kĩ thuật sinh thiết gai nhau (CVS).Từ mẫu mô thai nhi thu được, bác sĩ tiến hành nuôi cấy tế bào hoặc bạch cầu để xác định nồng độ axit sialic tự do.
Cuối cùng, xét nghiệm di truyền phân tử nhằm tìm kiếm các biến thể gây bệnh trên gen SLC17A5 có thể được sử dụng nhằm xác nhận kết quả chẩn đoán.
Điều trị
Hiện nay, bệnh rối loạn dự trữ axit sialic tự do vẫn chưa có phương pháp điều trị đặc hiệu. Do đó, quá trình điều trị chủ yếu tập trung vào giải quyết các triệu chứng cụ thể xuất hiện trên từng bệnh nhân. Đối với tình trạng co giật, bác sĩ kiểm soát theo các tiêu chuẩn chung, bao gồm sử dụng thuốc chống co giật.
Can thiệp sớm đóng vai trò rất quan trọng, giúp trẻ mắc bệnh phát huy tối đa tiềm năng của mình. Các biện pháp hỗ trợ hữu ích bao gồm giáo dục đặc biệt, vật lý trị liệu nhằm cải thiện sức mạnh và khả năng phối hợp vận động, trị liệu ngôn ngữ cùng nhiều dịch vụ y tế, xã hội khác.
Ngoài ra, tư vấn di truyền cũng được khuyến nghị cho người bệnh và các thành viên trong gia đình.
Dạng di truyền
Bệnh di truyền theo kiểu lặn trên nhiễm sắc thể thường, có nghĩa là cả hai bản sao của gen trong mỗi tế bào đều mang đột biến. Cha mẹ của một cá nhân mắc bệnh lặn trên nhiễm sắc thể thường mỗi người mang một bản sao của gen bị đột biến, nhưng họ thường không có dấu hiệu và triệu chứng của bệnh.

Nguồn: U.S. National Library of Medicine
Phòng ngừa
Do bệnh di truyền lặn, cha mẹ mang đột biến dị hợp nên gần như không có biểu hiện bệnh, do đó rất khó phát hiện cho đến khi sinh con. Để chủ động phòng ngừa, cha mẹ nên làm xét nghiệm sàng lọc gen lặn để chủ động cho tương lai của con. Các thành viên trong gia đình nên khám sức khỏe và tầm soát bệnh định kỳ nếu có thành viên mắc bệnh. Các cặp vợ chồng trước khi mang thai cần tư vấn và xét nghiệm di truyền đảm bảo sinh con khỏe mạnh.
Các tên gọi khác
- FSASD
- Sialic acid storage disease
References
- National Library of Medicine. Salla disease. Retrieved January 10, 2026 from https://www.ncbi.nlm.nih.gov/gtr/conditions/C1096903/
- NORD. Lysosomal Free Sialic Acid Storage Disorders. Retrieved January 10, 2026 from https://rarediseases.org/rare-diseases/lysosomal-free-sialic-acid-storage-disorders/
- OMIM. SALLA DISEASE; SD. Retrieved January 10, 2026 from https://omim.org/entry/604369
- MedlinePlus. Free sialic acid storage disorder. Retrieved January 10, 2026 from https://medlineplus.gov/genetics/condition/free-sialic-acid-storage-disorder/





