GM1 gangliosidosis (GM1 gangliosidosis) là bệnh di truyền làm chết tế bào thần kinh trong não và tủy sống.
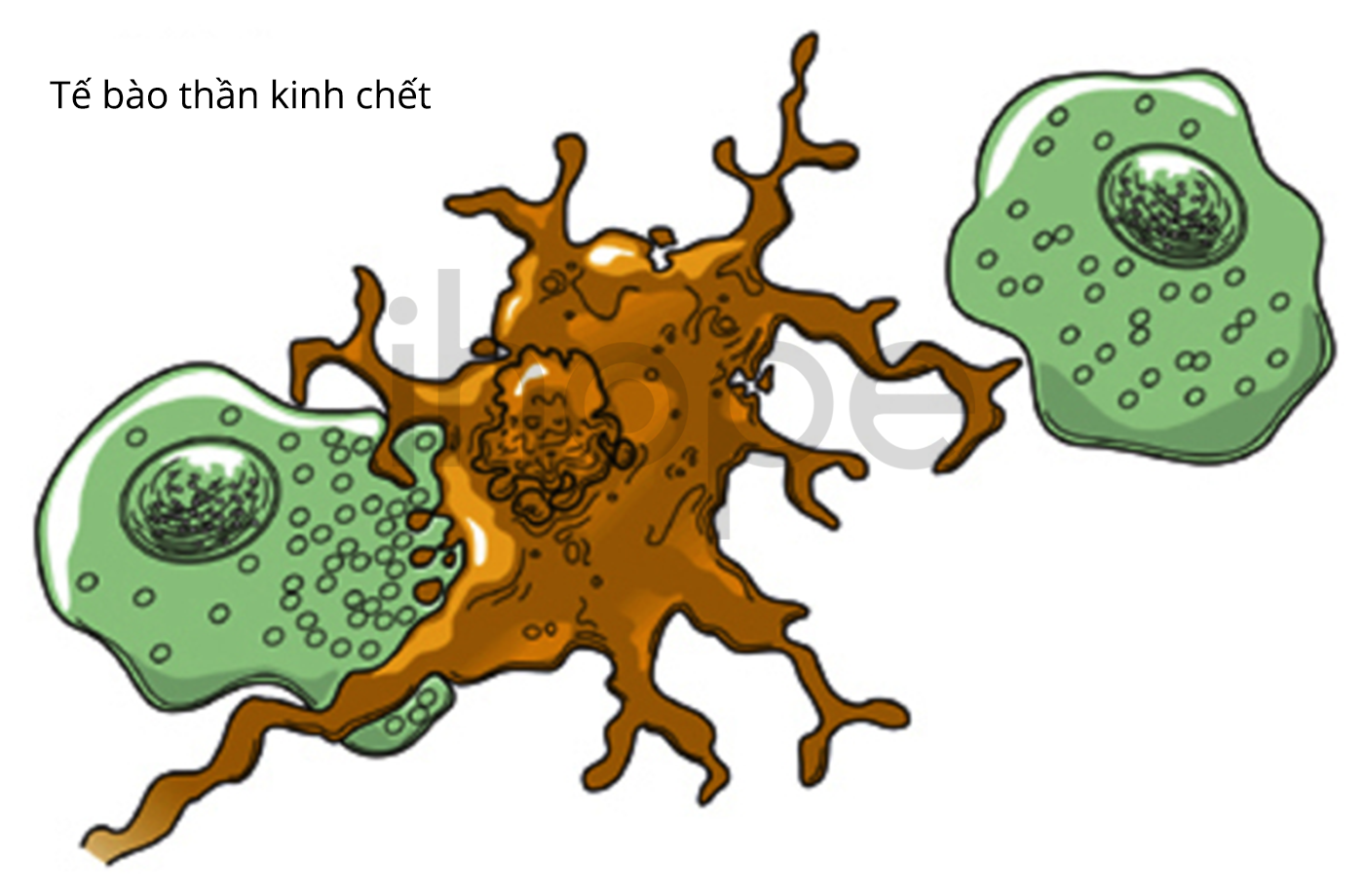
Tế bào đại thực bào (hai bên) tiêu thụ một tế bào thần kinh đang chết dần
Nguồn: U.S. National Library of Medicine
Biểu hiện lâm sàng
Bệnh GM1 gangliosidosis bao gồm ba loại chính tùy thuộc vào độ tuổi khởi phát bệnh. Tuy nhiên, triệu chứng của cả ba loại có nhiều điểm tương đồng dẫn đến khó phân biệt các dạng.
Loại I
GM1 gangliosidosis loại I là loại nghiêm trọng nhất. Thời điểm khởi phát bệnh phổ biến là khoảng 6 tháng tuổi. Trẻ sơ sinh mắc bệnh GM1 gangliosidosis loại I thường phát triển bình thường cho đến khi mức độ phát triển dần chậm lại và cơ vận động trở nên yếu đi. Mặt khác, trẻ mất dần các kĩ năng đã học trước đó và phản ứng quá mức với tiếng ồn lớn. Theo thời gian, gan và lách của người bệnh phình to. Đồng thời, bệnh nhân cũng xuất hiện các biểu hiện liên quan đến xương, co giật và thiểu năng trí tuệ mức độ nghiêm trọng.
Nguồn: Medlineplus.gov
Người bệnh GM1 gangliosidosis loại I còn có triệu chứng mất thị lực do lớp trong suốt bao phủ giác mạc trở nên mờ và mô cảm nhận ánh sáng của võng mạc tổn thương. Ngoài ra, mắt của bệnh nhân còn hình thành một vùng đỏ gọi là đốm đỏ anh đào. Trong một số trường hợp, bệnh nhân có đặc điểm khuôn mặt dị biệt bao gồm phì đại nướu răng , cơ tim phình to và yếu. Người bệnh thường không sống qua giai đoạn trẻ nhỏ.
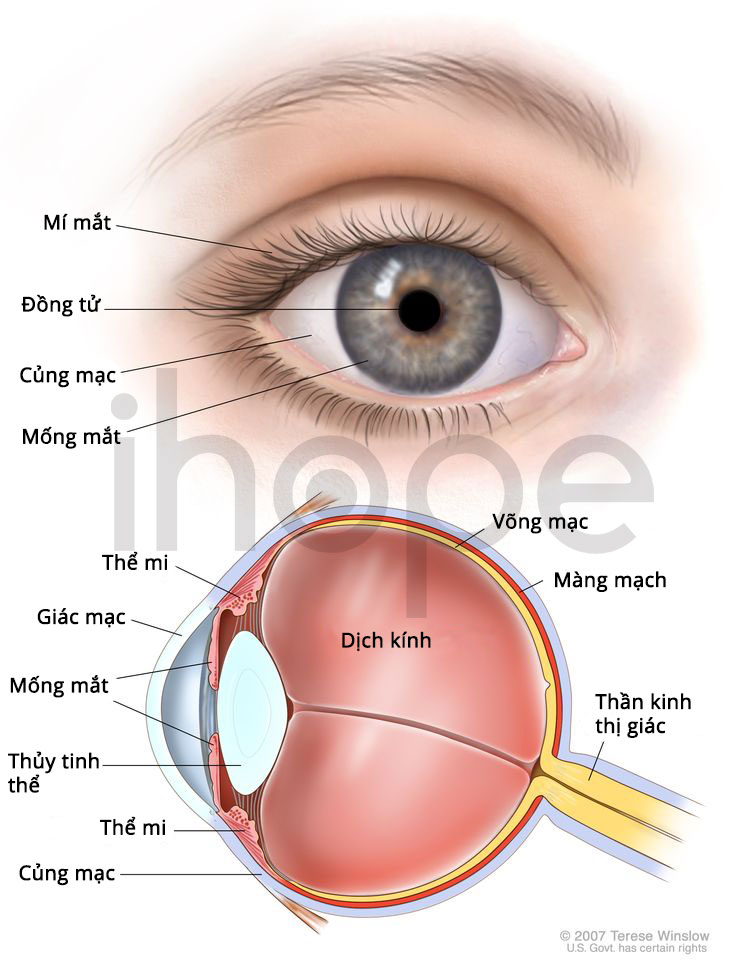
Nguồn: Medlineplus.gov
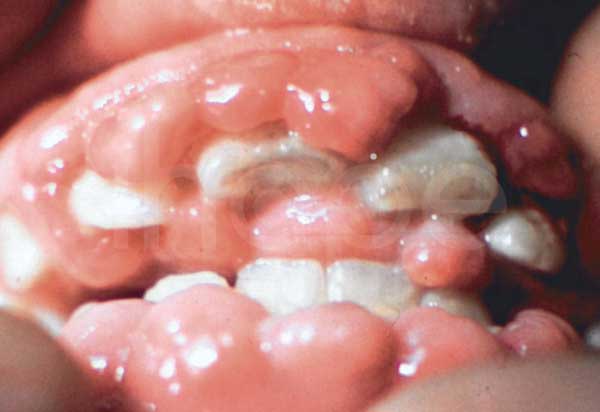
Ảnh: Nướu phì đại
Nguồn: Medlineplus.gov
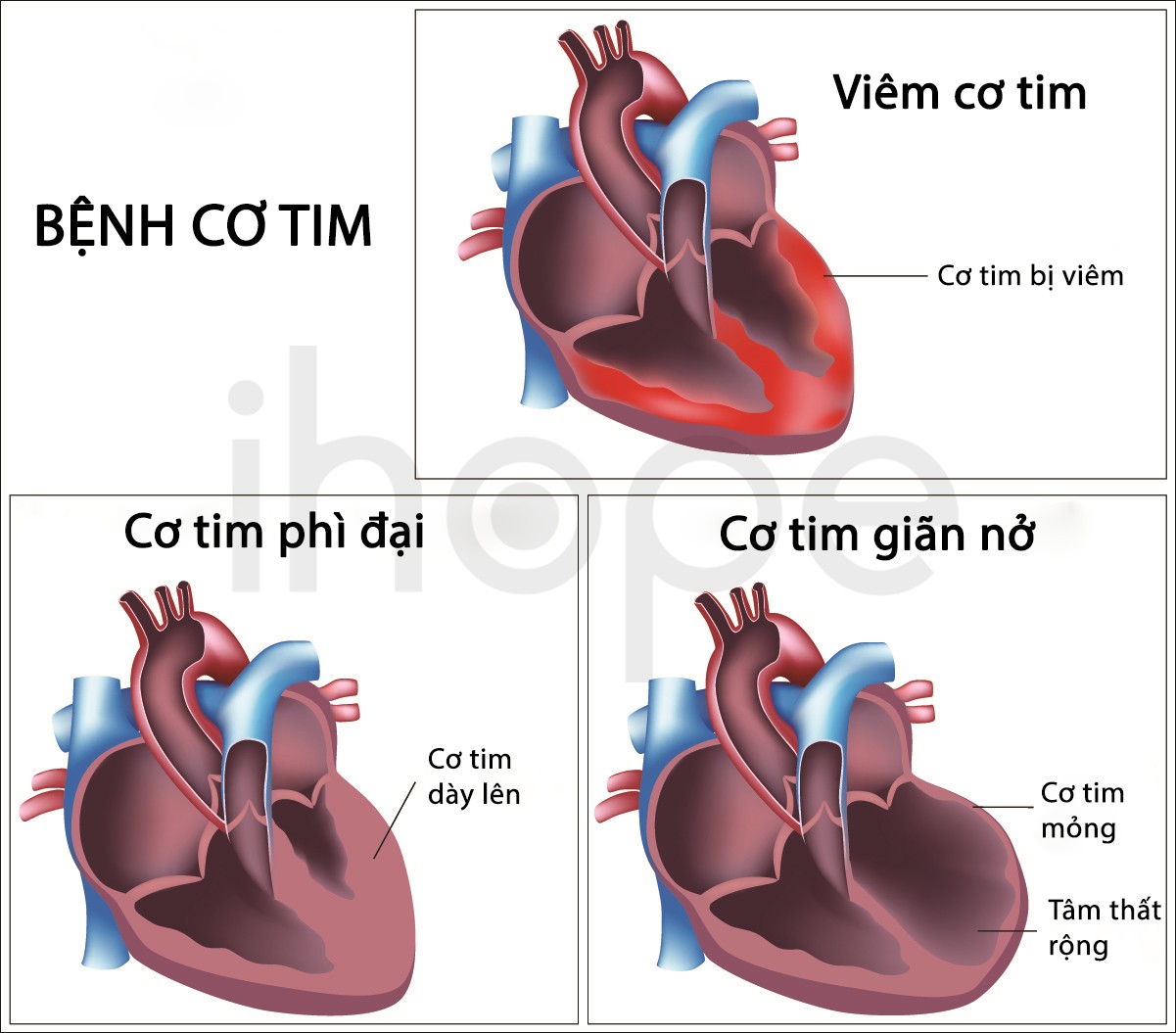
Ảnh: Bất thường của cơ tim
Nguồn: Alila Medical Media/Shutterstock.com
Loại II
GM1 gangliosidosis loại II gồm hai dạng:
- Dạng trẻ sơ sinh muộn: khởi phát khi trẻ 18 tháng tuổi
- Dạng thiếu niên: biểu hiện khi trẻ 5 tuổi
Bệnh nhân có dấu hiệu thoái triển về mặt phát triển nhưng không xuất hiện đốm đỏ anh đào, khuôn mặt thô hoặc cơ quan phình đại. Quá trình tiến triển của GM1 gangliosidosis loại II chậm hơn loại I nhưng vẫn làm giảm tuổi thọ người bệnh. Bệnh nhân thuộc dạng trẻ sơ sinh muộn thường sống đến giữa giai đoạn trẻ nhỏ, trong khi bệnh nhân thuộc dạng thiếu niên sống đến đầu thời kì trưởng thành.
Loại III
GM1 gangliosidosis loại III có mức độ nhẹ nhất. Bệnh thường xuất hiện trong độ tuổi trưởng thành và là bệnh mãn tính. Độ tuổi khởi phát bệnh khá đa dạng nhưng phần lớn người bệnh xuất hiện dấu hiệu vào độ tuổi thiếu niên. Những triệu chứng của loại này bao gồm loạn trương lực cơ và các dấu hiệu liên quan đến đốt sống. Tuổi thọ của người bệnh khác nhau giữa các trường hợp.
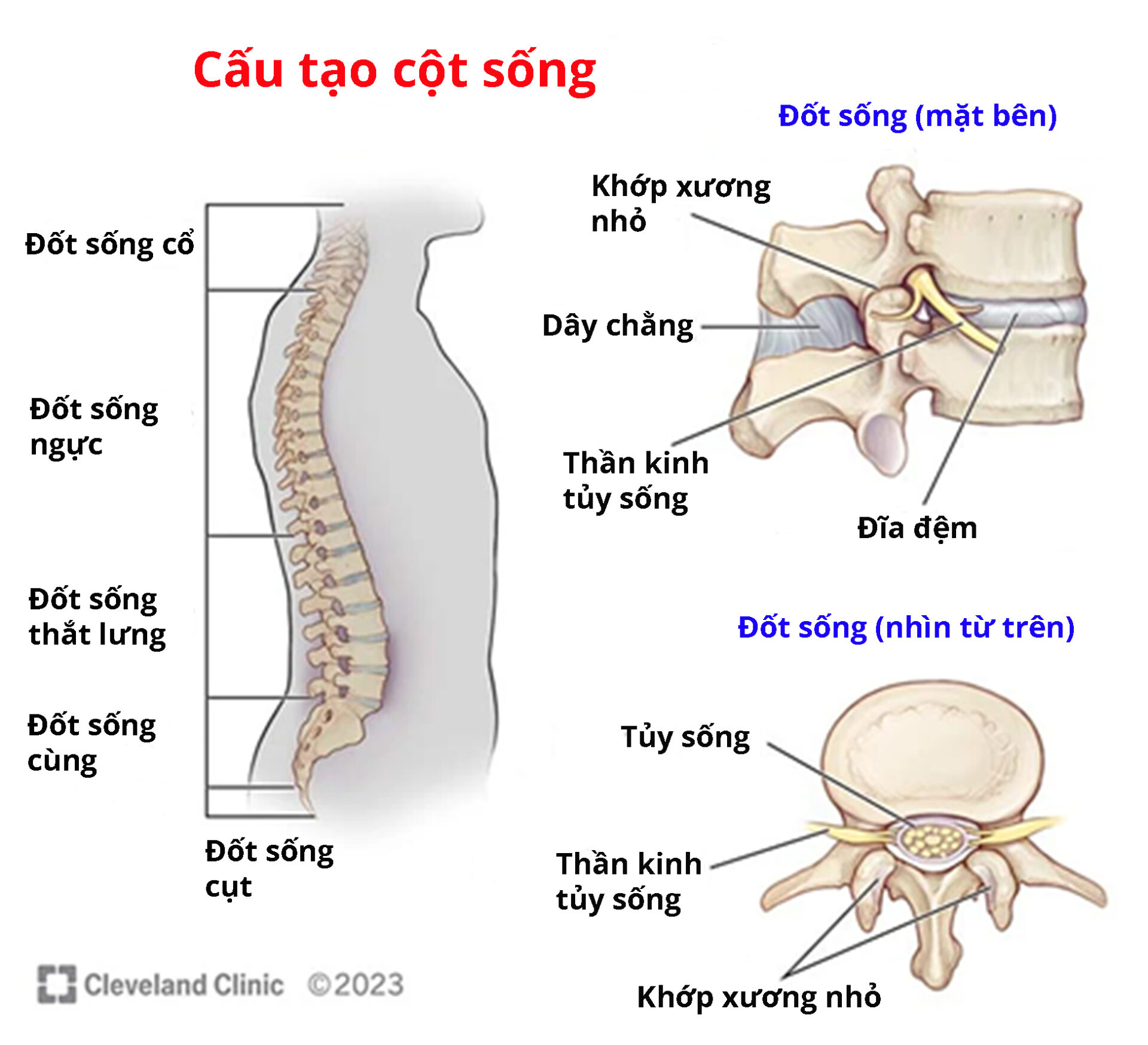
Nguồn: Cleverlandclinic.org
Độ phổ biến
Hiện nay, tỉ lệ mắc bệnh GM1 gangliosidosis vào khoảng 1/100.000–1/200.000 trẻ sơ sinh. Trong đó, loại I chiếm đa số trường hợp bệnh. Phần lớn người bệnh mắc loại III thường có gốc Nhật Bản.
Nguyên nhân
Đột biến gen GLB1 gây ra bệnh GM1 gangliosidosis. Gen GLB1 cung cấp hướng dẫn tạo ra enzyme beta-galactosidase tập trung trong lysosome. Lysosome có chức năng phân giải và tái sử dụng nhiều loại phân tử khác nhau. Enzyme beta-galactosidase hỗ trợ phân giải nhiều loại phân tử như cơ chất GM1 ganglioside. GM1 ganglioside duy trì hoạt động bình thường của tế bào thần kinh trong não.
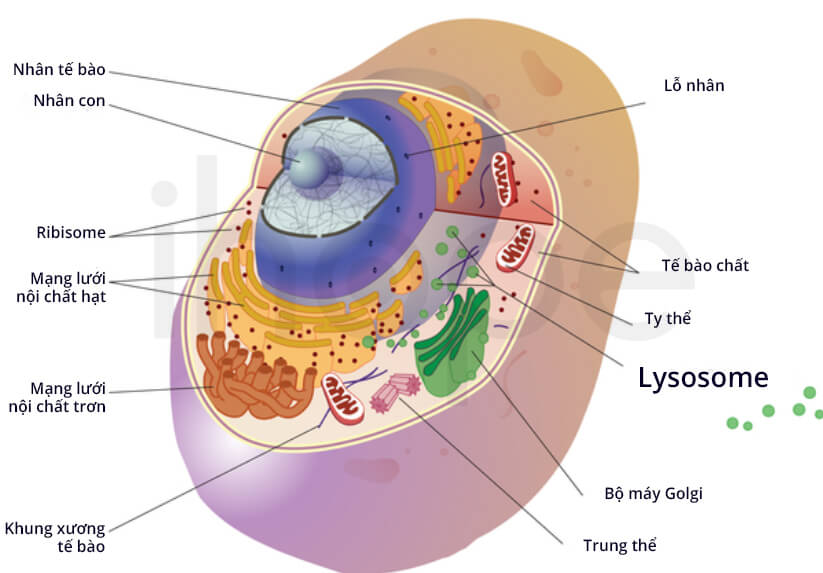
Nguồn: Darryl Leja, NHGRI
Đột biến gen GLB1 tạo ra enzyme beta-galactosidase không có khả năng phân giải GM1 ganglioside. Thiếu enzyme beta-galactosidase khiến GM1 ganglioside dư thừa tích lũy. Nồng độ GM1 ganglioside cao sẽ gây độc cho mô và cơ quan, chủ yếu là não. Tổn thương do GM1 ganglioside tích tụ dẫn đến cái chết của nhiều tế bào thần kinh và làm xuất hiện dấu hiệu bệnh.
Nhìn chung, người bệnh có triệu chứng nhẹ nếu enzyme beta-galactosidase vẫn còn hoạt động chút ít. Cơ thể người bệnh vẫn có khả năng phân giải một lượng GM1 ganglioside nhất định nên lượng enzyme tích trữ trong lysosome không nhiều.
Những bệnh giống GM1 gangliosidosis khiến các phân tử tích tụ trong lysosome được gọi là bệnh dự trữ lysosome.
Chẩn đoán
Trong trường hợp bệnh nhân có người thân mắc bệnh, sàng lọc trước sinh có thể chẩn đoán thay đổi di truyền xảy ra đối với thai nhi. Người mẹ nên thực hiện chọc ối hoặc lấy mẫu nhung mao màng đệm để xác định các biến đổi di truyền.
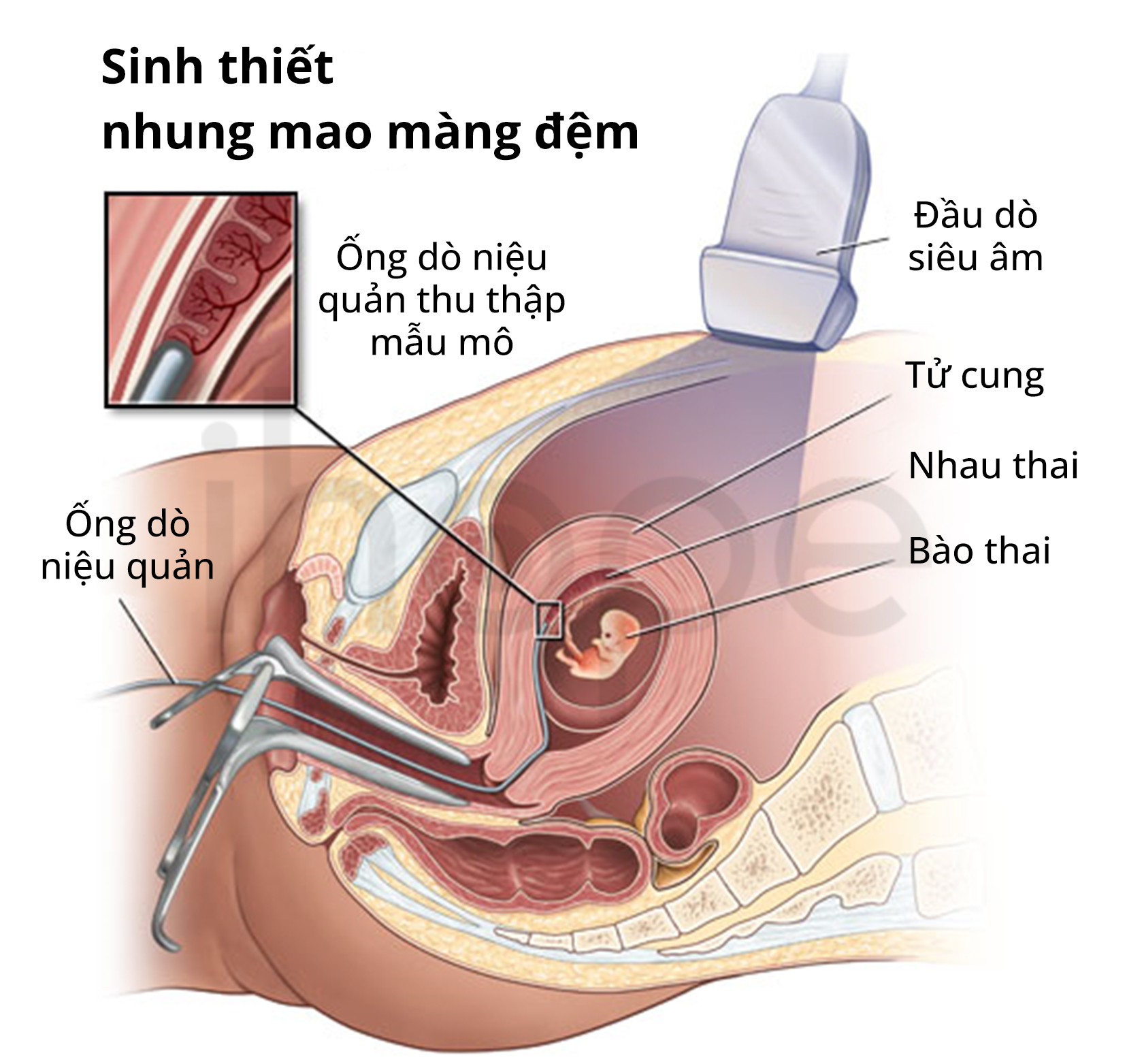
Nguồn: Johns Hopkins Medicine
Đối với trẻ sơ sinh và người lớn, bác sĩ thường chỉ định thực hiện các xét nghiệm sau:
- Xét nghiệm enzyme: xác định lượng beta-galactosidase trong máu
- Xét nghiệm di truyền: kiểm tra trình tự ADN nhằm xác định đột biến gen GLB1
- Khám sàng lọc trẻ sơ sinh: nhằm phát hiện các bệnh dự trữ lysosome
Điều trị
Hiện nay, chưa có liệu pháp điều trị bệnh GM1 gangliosidosis. Các liệu pháp tập trung làm giảm triệu chứng và cải thiện cuộc sống cho người bệnh. Bác sĩ thường kê thuốc chống co giật như gabapentin kết hợp với chế độ ăn keto nhằm hỗ trợ bệnh nhân giảm tần suất của các cơn động kinh.
Ngoài ra, còn có nhiều liệu pháp đang trong quá trình nghiên cứu và có tiềm năng áp dụng trong tương lai bao gồm liệu pháp tăng cường enzyme hoặc thay thế enzyme, liệu pháp gen, cấy ghép tế bào gốc và liệu pháp giảm cơ chất làm chậm tiến triển của bệnh.
Dạng di truyền
Bệnh GM1 gangliosidosis di truyền theo kiểu lặn trên nhiễm sắc thể thường, nghĩa là cả hai bản sao của gen trong mỗi tế bào đều mang đột biến. Cha mẹ của một cá nhân mắc bệnh lặn trên nhiễm sắc thể thường đều mang một bản sao của gen đột biến, nhưng họ thường không biểu hiện dấu hiệu và triệu chứng của bệnh.

Nguồn: U.S. National Library of Medicine
Phòng ngừa
Bệnh GM1 gangliosidosis di truyền lặn đột biến gen GLB1, cha mẹ mang đột biến dị hợp nên gần như không có biểu hiện bệnh, do đó rất khó phát hiện cho đến khi sinh con. Cha mẹ nên làm xét nghiệm sàng lọc gen lặn để chủ động cho tương lai của con. Các cặp vợ chồng trước khi mang thai cần tư vấn và xét nghiệm di truyền nhằm đảm bảo sinh con khỏe mạnh. Người thuộc nhóm nguy cơ mắc bệnh cần khám sức khỏe và tầm soát bệnh định kì.
Các tên gọi khác
- Beta-galactosidase-1 (GLB1) deficiency
References
- Genetic Testing Information. GM1 gangliosidosis. Retrieved September 04, 2024 from https://www.ncbi.nlm.nih.gov/gtr/conditions/C0085131/
- Genetic and Rare Diseases Information Center. GM1 gangliosidosis. Retrieved September 04, 2024 from https://rarediseases.info.nih.gov/diseases/10891/index
- Catalog of Genes and Diseases from OMIM. GM1-GANGLIOSIDOSIS, TYPE I; GM1G1. Retrieved September 04, 2024 from https://omim.org/entry/230500
- U.S. National Library of Medicine. GM1 gangliosidosis. Retrieved September 04, 2024 from https://medlineplus.gov/genetics/condition/gm1-gangliosidosis/
- Cleveland Clinic. GM1 Gangliosidosis. Retrieved September 04, 2024 from https://my.clevelandclinic.org/health/diseases/24069-gm1-gangliosidosis
- Frontiers. GM1 Gangliosidosis—A Mini-Review. Retrieved September 04, 2024 from https://www.frontiersin.org/journals/genetics/articles/10.3389/fgene.2021.734878/full
- National Institute of Health. GM1 Gangliosidosis: Mechanisms and Management. Retrieved September 04, 2024 from https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8044076/
- Orphanet. GM1 gangliosidosis. Retrieved September 04, 2024 from https://www.orpha.net/en/disease/detail/354
- MPS Society. GM1 Gangliosidosis. Retrieved September 04, 2024 from https://mpssociety.org.uk/conditions/related-conditions/gm1-gangliosidosis