
Thiếu acyl-CoA dehydrogenase chuỗi rất dài (Very Long-Chain Acyl-CoA Aehydrogenase Deficiency – VLCADD) là bệnh khiến cơ thể mất khả năng chuyển đổi một số chất béo thành năng lượng. Năng lượng này được cung cấp cho cơ thể sử dụng trong thời gian không ăn.
Biểu hiện lâm sàng
Thiếu acyl-CoA dehydrogenase chuỗi rất dài được phân thành ba dạng dựa trên thời gian khởi phát. Mỗi dạng có dấu hiệu, triệu chứng và mức độ nghiêm trọng khác nhau.
Đối với cả ba dạng, người bệnh có thể xuất hiện dấu hiệu bệnh khi nhịn ăn trong thời gian dài, bị nhiễm virus, tập thể dục và tiếp xúc với nhiệt độ quá nóng hoặc quá lạnh. Tuy nhiên, thiếu acyl-CoA dehydrogenase chuỗi rất dài thường bị nhầm lẫn với hội chứng Reye. Hội chứng Reye là bệnh hiếm gặp trên trẻ em, bệnh thường khởi phát sau khi trẻ bị nhiễm virus thủy đậu hoặc cúm.
Thiếu acyl-CoA dehydrogenase chuỗi rất dài dạng 1
Đây là dạng nghiêm trọng nhất và khởi phát sớm khi còn là trẻ sơ sinh. Trẻ có thể bị hạ đường huyết do lượng đường glucose trong máu thấp. Bên cạnh đó, trẻ nguy cơ gặp các biến chứng về gan và tim , dẫn đến đe dọa tính mạng.
Dấu hiệu và triệu chứng trong giai đoạn này bao gồm:
- Hôn mê
- Yếu cơ
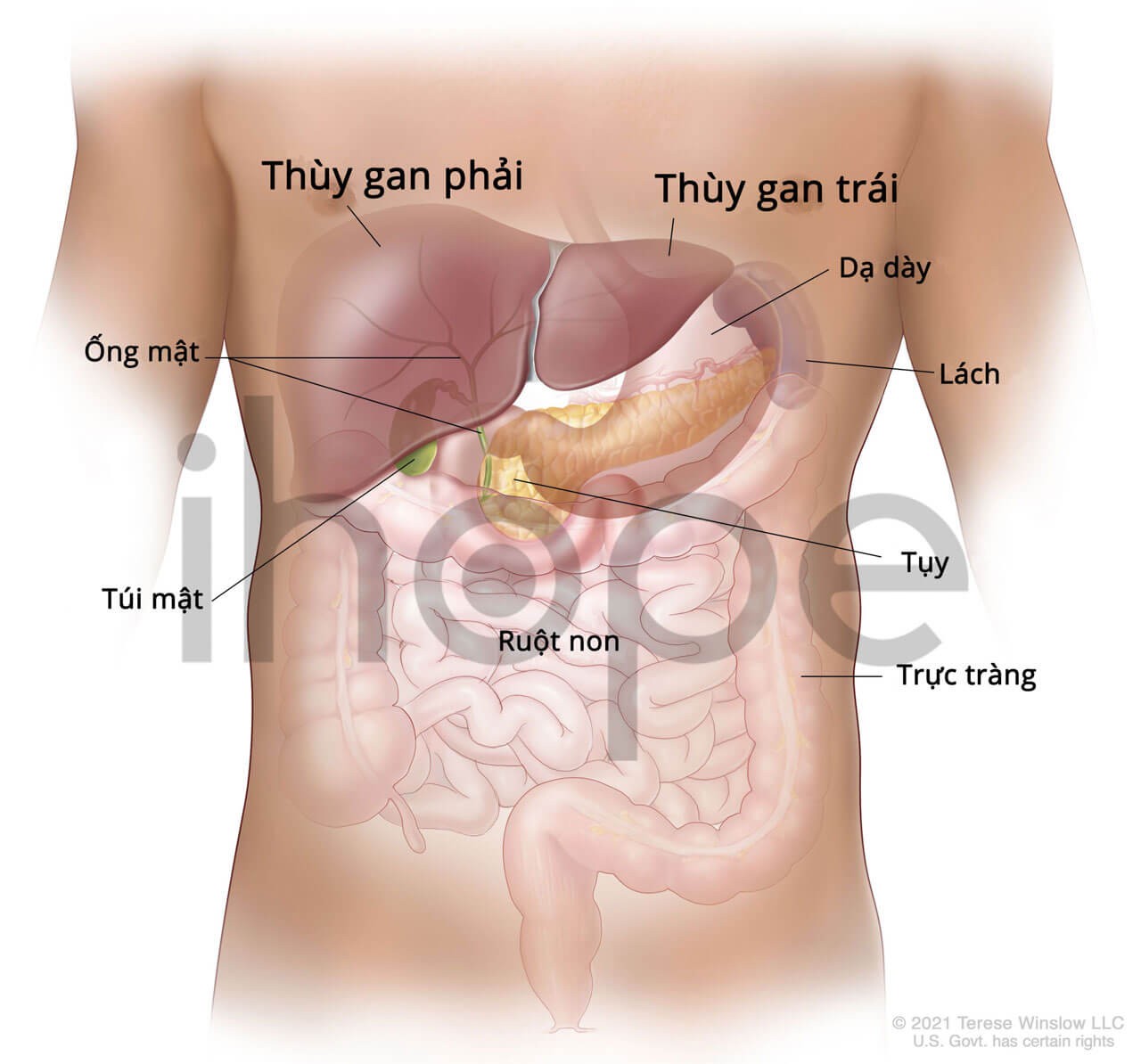
Ảnh: giải phẫu gan người
Nguồn: Terese Winslow LLC

Ảnh: Cấu trúc tim
Nguồn: American Heart Association
Thiếu acyl-CoA dehydrogenase chuỗi rất dài dạng 2
Thiếu acyl-CoA dehydrogenase chuỗi rất dài dạng 2 thường khởi phát trong giai đoạn trẻ nhỏ. Dạng này còn được gọi là hypoketotic hypoglycemic do người bệnh có mức độ axit béo ketone và lượng đường trong máu thấp hơn bình thường. Biểu hiện lâm sàng của người bệnh là gan to và đường huyết giảm. Ngoài ra, trẻ có thể gặp những bệnh khác liên quan đến gan hoặc bị yếu cơ.
Thiếu acyl-CoA dehydrogenase chuỗi rất dài dạng 3
Dạng 3 thường khởi phát khi người bệnh trưởng thành. Người bệnh xuất hiện triệu chứng đau cơ và tiêu cơ vân. Quá trình phá hủy mô cơ này sẽ giải phóng một lượng lớn protein myoglobin, sau đó chúng được thận xử lý và thải ra trong nước tiểu. Đây là hiện tượng myoglobin niệu, dẫn đến nước tiểu người bệnh có màu đỏ hoặc nâu.

Nguồn: Medlineplus.gov
Độ phổ biến
Theo thống kê, tỉ lệ mắc bệnh thiếu acyl-CoA dehydrogenase chuỗi rất dài khoảng 1/40.000–120.000 trường hợp.
Nguyên nhân
Đột biến gen ACADVL gây ra bệnh thiếu acyl-CoA dehydrogenase chuỗi rất dài. Gen này cung cấp hướng dẫn tạo ra enzyme acyl-CoA dehydrogenase chuỗi rất dài có chức năng chuyển hóa nhóm axit béo chuỗi rất dài. Nhóm axit béo này hiện diện trong nhiều loại thực phẩm cũng như các mô mỡ của cơ thể. Axit béo là nguồn cung cấp năng lượng chính cho hoạt động chức năng của tim và cơ bắp . Trong thời gian nhịn ăn, axit béo cũng là nguồn năng lượng quan trọng cho gan và nhiều mô khác duy trì hoạt động.
Đột biến gen ACADS ảnh hưởng đến quá trình sản xuất enzyme acyl-CoA dehydrogenase chuỗi rất dài. Enzyme này hoạt động trong ti thể—trung tâm sản xuất năng lượng của tế bào. Acyl-CoA dehydrogenase chuỗi rất dài cần thiết cho hoạt động oxy hóa axit béo—quá trình nhiều bước chuyển hóa chất béo thành năng lượng. Enzyme bị thiếu nên axit béo chuỗi rất dài không được chuyển hóa, dẫn đến cơ thể không có năng lượng để hoạt động. Do đó, người bệnh xuất hiện triệu chứng như mệt mỏi, hạ đường huyết và yếu cơ.
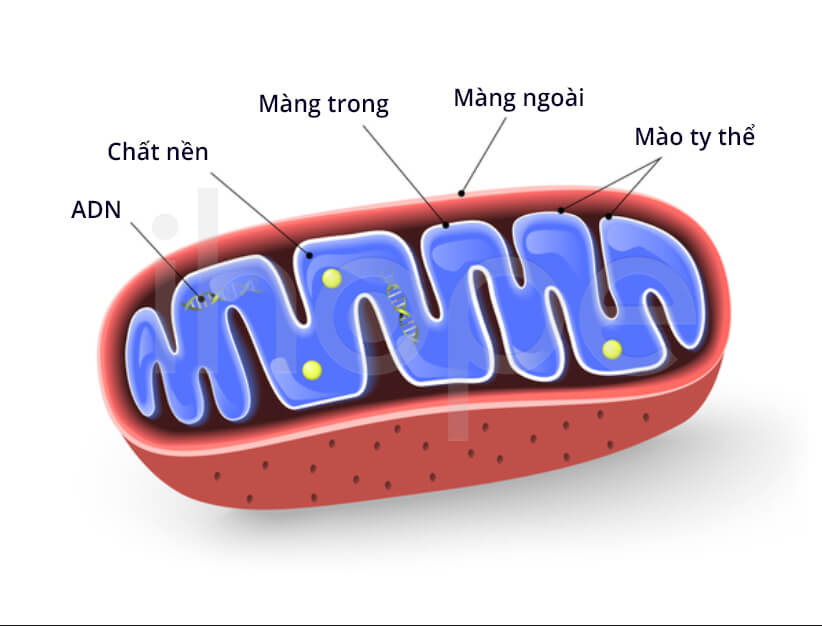
Nguồn: Designua/Shutterstock.com
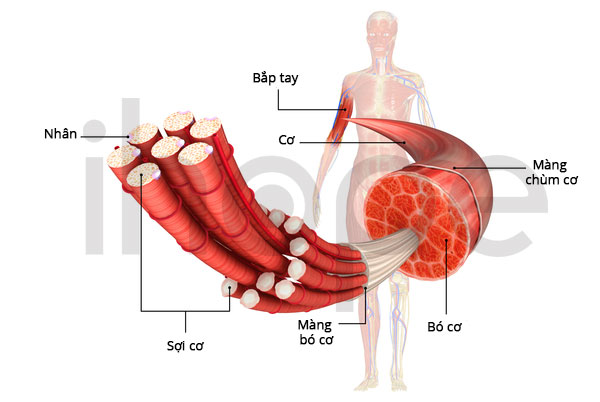
Ảnh: Giải phẫu cơ
Nguồn: Medlineplus.gov
Chẩn đoán
Thiếu acyl-CoA dehydrogenase chuỗi rất dài được chẩn đoán dựa trên biểu hiện lâm sàng, xét nghiệm chuyên biệt kết hợp với tiền sử bệnh gia đình. Ngoài ra, bác sĩ có thể chỉ định xét nghiệm di truyền nhằm xác nhận kết quả chẩn đoán.
Trong vòng 24–48 giờ sau khi sinh, trẻ được lấy máu gót chân để thực hiện sàng lọc sơ sinh nhằm phát hiện tình trạng thiếu acyl-CoA dehydrogenase chuỗi rất dài và các rối loạn khác. Nếu kết quả cho thấy nguy cơ cao mắc bệnh, bác sĩ sẽ cho em bé làm thêm xét nghiệm chẩn đoán. Điều quan trọng cần lưu ý là kết quả sàng lọc nguy cơ cao không đồng nghĩa với kết luận em bé mắc bệnh. Kết quả này có thể do mẫu máu ban đầu được thu quá ít hoặc quá sớm. Tuy nhiên, cha mẹ nên đưa trẻ tái khám để làm xét nghiệm xác nhận. Nếu không được điều trị, bệnh có thể ảnh hưởng đến sức khỏe của trẻ rõ rệt ngay sau khi sinh, xét nghiệm tiếp theo phải được tiến hành càng sớm càng tốt để xác định xem liệu trẻ có mắc bệnh hay không.
Xét nghiệm tiếp theo bao gồm kiểm tra mẫu nước tiểu và máu của bé để đo nồng độ axit và chất độc có hại. Một số axit và độc tố tích tụ trong cơ thể khi trẻ bị rối loạn quá trình oxy hóa axit béo, do đó, bác sĩ có thể dựa vào nồng độ các chất này để xác định xem em bé có mắc bệnh hay không. Nồng độ cao acylcarnitine C14:1 trong máu có thể là dấu hiệu của bệnh thiếu acyl-CoA dehydrogenase chuỗi rất dài. Đôi khi, bác sĩ cần xét nghiệm thêm một mẫu da rất nhỏ.
Điều trị
Hiện nay, thiếu acyl-CoA dehydrogenase chuỗi rất dài có thể được điều trị nâng đỡ bằng nhiều phương pháp.
Ăn uống đầy đủ
Một số trẻ sơ sinh và trẻ nhỏ bị thiếu acyl-CoA dehydrogenase chuỗi rất dài cần ăn thường xuyên để ngăn ngừa khủng hoảng trao đổi chất. Bác sĩ sẽ cho biết tần suất trẻ cần được cho ăn. Nhìn chung, trẻ sơ sinh nên được cho ăn cứ bốn đến sáu giờ một lần, một số bé thậm chí cần ăn thường xuyên hơn. Điều quan trọng là trẻ sơ sinh được cho ăn vào ban đêm. Trẻ có thể cần được đánh thức để ăn nếu không tự thức dậy. Bác sĩ hoặc chuyên gia dinh dưỡng sẽ hướng dẫn cha mẹ thực hiện kế hoạch ăn uống phù hợp cho trẻ sơ sinh, đặc biệt khi bé bị bệnh hoặc khi bé không chịu ăn.
Bác sĩ trao sẽ tư vấn chế độ ăn khi bé lớn hơn. Khi khỏe mạnh, nhiều thanh thiếu niên và người lớn có thể nhịn ăn tới 12 giờ mà không gặp vấn đề gì. Các phương pháp điều trị khác nên được tiếp tục suốt cuộc đời.
Chế độ ăn phù hợp
Đôi khi, trẻ mắc bệnh nên áp dụng chế độ ăn ít chất béo và nhiều carbohydrate. Carbohydrate cung cấp cho cơ thể nhiều loại đường có thể được sử dụng làm năng lượng. Trên thực tế, trẻ nên ăn nhiều loại thực phẩm giàu carbohydrate (bánh mì, mì ống, trái cây, rau củ,...) và protein (thịt nạc và thực phẩm từ sữa ít béo). Cha mẹ nên trao đổi với bác sĩ trước khi thực hiện bất kỳ thay đổi với chế độ ăn uống.
Người bệnh không thể sử dụng một số loại chất béo được gọi là axit béo chuỗi dài. Phần lớn chất béo còn lại trong chế độ ăn có thể dưới dạng axit béo chuỗi trung bình.
Triglyceride chuỗi trung bình
Triglyceride chuỗi trung bình (Medium Chain Triglyceride - MCT) thường được đưa vào chế độ ăn dành cho những người mắc bệnh thiếu acyl-CoA dehydrogenase chuỗi rất dài. Triglyceride chuỗi trung bình có sẵn trong dầu triglyceride chuỗi trung bình và triheptanoin (DOJOLVI®)—một loại thuốc làm từ triglyceride chuỗi trung bình. Những loại thực phẩm bổ sung này chứa axit béo chuỗi trung bình có thể được sử dụng với số lượng nhỏ để tạo năng lượng. Bác sĩ sẽ tư vấn cách sử dụng và liều lượng phù hợp cho mỗi bệnh nhân.
L-Carnitine và các chất bổ sung khác
Một số trẻ có thể cần bổ sung L-carnitine—chất an toàn và tự nhiên giúp cơ thể tạo ra năng lượng. L-carnitine cũng giúp cơ thể loại bỏ các chất thải có hại. Bác sĩ sẽ quyết định xem em b1 có cần L-carnitine hay không.
Các chất bổ sung khác có thể được khuyên dùng cho các trường hợp trẻ sơ sinh mắc bệnh nặng. Lưu ý không sử dụng bất kỳ loại thuốc hoặc chất bổ sung nào mà chưa hỏi ý kiến bác sĩ.
Gặp bác sĩ khi có triệu chứng
Cha mẹ nên đưa con đến gặp bác sĩ nếu phát hiện bé có bất kỳ biểu hiện sau đây:
- Ăn kém
- Mệt mỏi hoặc buồn ngủ quá mức
- Nôn mửa
- Tiêu chảy
- Nhiễm trùng
- Sốt
- Đau hoặc yếu cơ dai dẳng, nước tiểu có màu nâu đỏ
Trẻ cần ăn thêm thực phẩm giàu tinh bột và uống nhiều nước hơn ngay cả khi không cảm thấy đói để phòng ngừa hạ đường huyết. Trẻ bệnh thường không muốn ăn. Nếu không thể ăn, trẻ có thể cần được điều trị tại bệnh viện để ngăn ngừa biến chứng.
Tránh tập thể dục hoặc gắng sức kéo dài
Tập thể dục nặng trong thời gian dài cũng có thể gây ra các triệu chứng như sau:
- Đau cơ
- Chuột rút
- Yếu ớt
- Nước tiểu màu nâu đỏ
Nếu các triệu chứng về cơ xảy ra, người bệnh cần điều trị kịp thời để ngăn ngừa tổn thương thận. Trẻ em hoặc người lớn có triệu chứng về cơ nên:
- Uống nước ngay lập tức
- Ăn tinh bột hoặc đường
- Đến bệnh viện để điều trị
Để ngăn ngừa các triệu chứng về cơ, hãy thực hiện:
- Tránh tập thể dục kéo dài hoặc nặng
- Giữ ấm cơ thể
- Ăn carbohydrate trước và trong thời gian tập thể dục vừa phải
Dạng di truyền
Thiếu acyl-CoA dehydrogenase chuỗi rất dài di truyền theo kiểu lặn trên nhiễm sắc thể thường. Do đó, bệnh chỉ biểu hiện khi có cả hai bản sao của gen đột biến trong mỗi tế bào. Bệnh nhân mắc bệnh lặn trên nhiễm sắc thể thường sẽ có bố và mẹ mang một bản sao của gen đột biến, nhưng bố mẹ ít khi biểu hiện triệu chứng bệnh.

Nguồn: U.S. National Library of Medicine
Phòng ngừa
Thiếu acyl-CoA dehydrogenase chuỗi rất dài di truyền lặn bắt nguồn từ đột biến gen ACADVL, cha mẹ mang đột biến dị hợp nên gần như không có biểu hiện bệnh, do đó rất khó phát hiện cho đến khi sinh con. Để chủ động phòng ngừa, cha mẹ nên làm xét nghiệm sàng lọc gen lặn để chủ động cho tương lai của con. Các cặp vợ chồng trước khi mang thai cần tư vấn và xét nghiệm di truyền đảm bảo sinh con khỏe mạnh. Người thuộc nhóm nguy cơ mắc bệnh cần khám sức khỏe và tầm soát bệnh định kỳ.
Các tên gọi khác
- ACADVL
- Acyl-CoA dehydrogenase very long chain deficiency
- Very long-chain acyl coenzyme A dehydrogenase deficiency
- Very long-chain acyl-coenzyme A dehydrogenase deficiency
- VLCAD deficiency
- VLCAD-C
- VLCAD-H
References
- Genetic Testing Information. Very long chain acyl-CoA dehydrogenase deficiency. Retrieved September 27, 2023 from https://www.ncbi.nlm.nih.gov/gtr/conditions/C3887523/
- Genetic and Rare Diseases Information Center. Very long chain acyl-CoA dehydrogenase deficiency. Retrieved September 27, 2023 from https://rarediseases.info.nih.gov/diseases/5508/vlcad-deficiency
- Catalog of Genes and Diseases from OMIM. ACYL-CoA DEHYDROGENASE, VERY LONG-CHAIN, DEFICIENCY OF; ACADVLD. Retrieved September 27, 2023 from https://omim.org/entry/201475
- U.S National Library of Medicine. Very long-chain acyl-CoA dehydrogenase deficiency. Retrieved 27 September 27, 2023 from https://medlineplus.gov/genetics/condition/very-long-chain-acyl-coa-dehydrogenase-deficiency/
- Baby's First Test. Very Long-Chain Acyl-CoA Dehydrogenase Deficiency. Retrieved October 2, 2023 from https://www.babysfirsttest.org/newborn-screening/conditions/very-long-chain-acyl-coa-dehydrogenase-deficiency
- Health Resources and Services Administration Very long-chain acyl-CoA dehydrogenase deficiency. Retrieved September 27, 2023 from https://newbornscreening.hrsa.gov/conditions/very-long-chain-acyl-coa-dehydrogenase-deficiency
- Mayo Clinic. Very Long Chain Acyl-CoA Dehydrogenase Deficiency, Full Gene Analysis, Varies. Retrieved September 27, 2023 from https://www.mayocliniclabs.com/test-catalog/Overview/35571
- National Organization for Rare Disorders. Very Long Chain Acyl CoA Dehydrogenase Deficiency (LCAD). Retrieved September 27, 2023 from https://rarediseases.org/rare-diseases/very-long-chain-acyl-coa-dehydrogenase-deficiency-lcad/
- Orphanet. Very long chain acyl-CoA dehydrogenase deficiency. Retrieved September 27, 2023 from https://www.orpha.net/consor/www/cgi-bin/OC_Exp.php?lng=EN&Expert=26793
- STAR-G. VLCADD (very long chain acyl-CoA dehydrogenase deficiency). Retrieved September 27, 2023 from https://www.newbornscreening.info/vlcadd-very-long-chain-acyl-coa-dehydrogenase-deficiency/#13





