
Bệnh xương thủy tinh hay tạo xương bất toàn (Osteogenesis Imperfecta – OI) là một nhóm các rối loạn di truyền chủ yếu ảnh hưởng đến xương. Thuật ngữ “tạo xương bất toàn” chỉ quá trình hình thành xương không toàn vẹn. Người bệnh dễ bị gãy xương khi gặp chấn thương nhẹ hoặc không rõ nguyên nhân. Nhiều trường hợp gãy xương phổ biến, trong những trường hợp nghiêm trọng, gãy xương có thể xảy ra ngay cả trước khi sinh. Các trường hợp nhẹ hơn có thể chỉ liên quan đến một vài ca gãy xương trong suốt cuộc đời người bệnh.
Bốn loại bệnh xương thủy tinh chính (loại collagen) được xác định dựa trên các đặc điểm lâm sàng và mức độ nghiêm trọng. Những loại này chiếm 85-90% các trường hợp và do đột biến gen COL1A1 hoặc COL1A2 gây ra. Các gen này mã hóa collagen loại 1 – loại collagen dồi dào nhất trong cơ thể con người. Nó được tìm thấy trong xương, gân và dây chằng. Xương thủy tinh loại I là dạng bệnh phổ biến nhất và nhẹ nhất. Xương thủy tinh loại II là dạng nghiêm trọng nhất trong số các loại collagen. Các loại bệnh từ V đến XXI (loại không phải collagen), cũng như các loại chưa được phân loại chiếm 10-15% các trường hợp xương thủy tinh. Những loại này là do những thay đổi trong gen mã hóa protein tương tác với collagen.
Biểu hiện lâm sàng
Trong tất cả loại bệnh xương thủy tinh, các triệu chứng khác nhau rất nhiều từ ở từng trường hợp ngay cả trong cùng một loại và cùng một gia đình. Một số người bệnh không bị gãy xương hoặc chỉ số ít, trong khi những người khác trải qua nhiều lần gãy xương. Tuổi bắt đầu gãy xương ở mỗi người khác nhau. Xương thủy tinh là bệnh liên quan đến collagen, do đó bệnh còn có thể ảnh hưởng đến răng, chức năng phổi, chức năng tim, sức mạnh cơ bắp và tính linh hoạt của dây chằng.
Trước đây, xương thủy tinh được phân thành bốn loại chính dựa trên đặc điểm lâm sàng và mức độ nghiêm trọng. Trong thập kỷ qua, người ta đã phát hiện thêm nhiều gen mới góp phần gây ra bệnh, do đó quá trình phân loại đã được mở rộng ra ngoài loại I đến IV để bao gồm các loại mới và hiếm gặp hơn. Các loại V đến XXI được phân theo đột biến gen gây bệnh. Tương tự như các loại viêm khớp hay gặp, người mắc loại bệnh xương thủy tinh hiếm gặp này có những đặc điểm lâm sàng khác nhau.
Loại I
Xương thủy tinh loại I là dạng phổ biến nhất và thường là dạng nhẹ nhất. Phần lớn người bệnh bị gãy nhiều xương, thường xảy ra từ thời thơ ấu đến tuổi dậy thì. Trẻ có thể bị gãy xương sớm chỉ với chấn thương nhẹ (ngã từ tư thế đứng hoặc khi được người chăm sóc kéo lên), trong khi những trẻ khác có thể bị gãy xương khi tham gia hoạt động thể chất cường độ cao. Hiếm khi gãy xương xảy ra trong thời kỳ sơ sinh. Tần suất gãy xương thường giảm sau tuổi dậy thì. Gãy xương nhiều lần có thể dẫn đến biến dạng nhẹ xương cánh tay và chân (ví dụ: cong xương chày và xương đùi).
Một đặc điểm phân biệt xương thủy tinh loại I là lòng trắng của mắt đổi màu hơi xanh (màng cứng xanh). Một số người có thể phát triển các bất thường ảnh hưởng đến tai giữa và/hoặc tai trong, góp phần hoặc dẫn đến suy giảm thính lực. Tỷ lệ mất thính lực tăng lên theo tuổi.
Người bệnh xương thủy tinh loại I có khuôn mặt hình tam giác. Chiều cao có thể thay đổi và hầu hết mọi người đều có chiều cao dưới trung bình theo độ tuổi khi còn nhỏ, chiều cao khi trưởng thành thấp hơn các thành viên khỏe mạnh trong gia đình. Từ 10-40% bệnh nhân phát triển chứng vẹo cột sống. Độ cong thường nhẹ và tiến triển chậm theo thời gian.
Các triệu chứng khác bao gồm khớp lỏng lẻo và yếu cơ có thể dẫn đến trật khớp và bong gân dây chằng. Một số bệnh nhân có da dễ bị bầm tím. Răng giòn (dentinogenesis) không phổ biến ở xương thủy tinh loại I.
Loại II
Xương thủy tinh loại II là dạng bệnh nghiêm trọng nhất. Trẻ sơ sinh thường gặp các biến chứng đe dọa tính mạng khi sinh hoặc ngay sau đó. Trẻ sinh ra nhẹ cân, tay chân ngắn bất thường và màng cứng màu xanh. Ngoài ra, trẻ có xương cực kỳ mỏng manh và nhiều vết gãy ngay từ khi chào đời. Xương sườn và xương dài của chân thường bị dị dạng.
Trẻ sơ sinh có phổi kém phát triển và lồng ngực nhỏ bất thường, có thể dẫn đến suy hô hấp đe dọa tính mạng. Một số trẻ bị suy tim sung huyết.
Trẻ sơ sinh cũng có thể có mũi nhỏ, hẹp, hàm nhỏ (micrognathia) và các nốt mềm lớn bất thường trên đỉnh hộp sọ (thóp lớn). Trẻ có da mỏng, dễ vỡ và yếu cơ.
Loại III
Bệnh xương thủy tinh loại III có biểu hiện xương dị dạng cực kỳ dễ gãy và nhiều vết gãy. Gãy xương thường xảy ra khi mới sinh, chụp X-quang có thể được dấu hiệu xương gãy rồi lành khi còn trong bụng mẹ.
Dị tật tiến triển của các xương khác nhau thường dẫn đến tầm vóc thấp, biến dạng cột sống (vẹo cột sống, vẹo ngực và cong vẹo thắt lưng) và dị dạng điểm nối xương phía sau hộp sọ (xương chẩm) và đỉnh cột sống. Khoảng 70% trẻ mắc bệnh xương thủy tinh loại III phát triển chứng vẹo cột sống. Các biến dạng thành ngực thường gặp dẫn đến lồng ngực hình thùng. Tình trạng chi trên và chi dưới thường xuyên gãy và biến dạng có thể phải phẫu thuật nhiều lần để ổn định khi trẻ lớn lên. Chiều cao khi trưởng thành bị giảm sút nghiêm trọng. Người bệnh phụ thuộc nhiều hơn vào xe lăn và các thiết bị hỗ trợ di chuyển khác ở độ tuổi thanh niên.
Trẻ sơ sinh có lòng trắng mắt một chút màu xanh lam khi mới sinh, sau đó nhạt dần trong năm đầu tiên. Trẻ thường có khuôn mặt hình tam giác do trán dô bất thường và hàm nhỏ (micrognathia). Mất thính giác có thể phát triển trong 10 năm đầu tiên. Bệnh nhân có thể phát triển các vấn đề về phổi thứ phát do bất thường mô phổi và bất thường thành ngực.
Loại IV
Mức độ nghiêm trọng của bệnh xương thủy tinh loại IV (loại trung bình) có thể giống loại I hoặc loại III. Gãy xương thường xảy ra trước tuổi dậy thì. Người bệnh bị dị dạng xương từ nhẹ đến trung bình và thường ngắn hơn bình thường. Bệnh nhân cũng có thể bị cong vẹo cột sống.
Người bệnh xương thủy tinh loại IV có khuôn mặt hình tam giác. Phần lớn bệnh nhân có màng cứng bình thường hoặc màu xanh nhạt trong thời kỳ sơ sinh. Khi trẻ lớn hơn, màu xanh nhạt của màng cứng sẽ mất dần. Người bệnh cũng có thể bị suy giảm thính lực và khiếm khuyết phát sinh cơ (dentinogenesis imperfecta).
Độ phổ biến
Bệnh xương thủy tinh ảnh hưởng đến khoảng 1/10.000 đến 1/20.000 người trên toàn thế giới. Ước tính có khoảng 25.000 đến 50.000 người bệnh ở Hoa Kỳ.
Nguyên nhân
Bệnh xương thủy tinh xảy ra do đột biến tại một trong số các gen liên quan. Đột biến gen COL1A1 và COL1A2 gây ra khoảng 90% các trường hợp. Những gen này cung cấp hướng dẫn tạo ra các protein được sử dụng để lắp ráp collagen loại I - loại protein phong phú nhất trong xương, da và các mô liên kết khác, nó cung cấp cấu trúc và sức mạnh cho cơ thể.
Bệnh xương thủy tinh loại I do đột biến gen COL1A1 hoặc ít phổ biến hơn là gen COL1A2. Những thay đổi di truyền này làm giảm lượng collagen loại I được sản xuất trong cơ thể, mặc dù chất lượng của chúng bình thường. Giảm collagen loại I làm cho xương giòn và dễ gãy. Các đột biến gây ra xương thủy tinh loại II, III và IV xảy ra tại gen COL1A1 hoặc COL1A2. Chúng thường làm thay đổi cấu trúc của các phân tử collagen loại I, dẫn đến collagen loại I bất thường. Một khiếm khuyết trong cấu trúc của collagen loại I làm suy yếu các mô liên kết, đặc biệt là xương, từ đó dẫn đến các khiếm khuyết nghiêm trọng hơn của quá trình tạo xương.
Đột biến tại các gen khác gây ra các dạng khiếm khuyết hiếm gặp của quá trình tạo xương. Nhiều gen trong số này hướng dẫn các protein giúp xử lý collagen loại I thành dạng trưởng thành, do đó khi bị đột biến, chúng làm gián đoạn các bước khác nhau trong quá trình sản xuất phân tử collagen. Những thay đổi này làm suy yếu các mô liên kết, dẫn đến bất thường nghiêm trọng về xương và vấn đề tăng trưởng. Các gen liên quan khác cung cấp hướng dẫn tạo ra protein kiểm soát sự phát triển và chức năng của tế bào tạo xương, vì vậy khi bị đột biến, chúng làm giảm sự phát triển bình thường của xương, khiến xương giòn và dễ gãy.
Chẩn đoán
Bệnh xương thủy tinh thường được di truyền từ cha hoặc mẹ mang bệnh. Chẩn đoán dựa trên tiền sử gia đình và/hoặc biểu hiện lâm sàng như gãy xương thường xuyên, tầm vóc thấp bé, lòng trắng mắt có màu xanh lam (màng cứng màu xanh), các vấn đề về răng và mất thính giác tiến triển sau tuổi dậy thì.
Chụp X-quang cũng được sử dụng để chẩn đoán xương thủy tinh. Phương pháp này có thể phát hiện gãy xương trong các giai đoạn lành khác nhau, một mẫu xương sọ bất ngờ được gọi là xương Wormian và xương ở cột sống được gọi là "đốt sống cá tuyết."
Các loại xét nghiệm bao gồm xét nghiệm sinh hóa hoặc giải trình tự gen COL1A1 và COL1A2. Xét nghiệm sinh hóa nghiên cứu các ảnh ghép lấy từ mẫu sinh thiết da nhỏ. Nếu collagen loại I thay đổi, đó chính là một dấu hiệu của bệnh. Trình tự ADN của COL1A1 và COL1A2 được sử dụng để xác định đột biến collagen loại I khiến gây ra protein collagen bị thay đổi. Xét nghiệm ADN cần lấy mẫu máu để tách chiết ADN. Cả hai xét nghiệm đều tương đối nhạy, phát hiện lần lượt khoảng 90% và 95% những người đã có chẩn đoán lâm sàng. Xét nghiệm di truyền và sinh hóa cho trẻ mắc bệnh xương thủy tinh có thể phát hiện các gen collagen ít phổ biến hơn (CRTAP và P3H (LEPRE1)) chịu trách nhiệm cho một số dạng lặn hiếm gặp của bệnh.
Điều trị
Hiện chưa có cách chữa khỏi bệnh xương thủy tinh mà chủ yếu liệu pháp hỗ trợ giảm gãy xương và khuyết tật, giúp bệnh nhân sống độc lập và duy trì sức khỏe chung. Cần có nhóm chuyên gia y tế bao gồm bác sĩ, thuốc, chỉnh hình và phục hồi chức năng. Liệu pháp hỗ trợ cho mỗi cá nhân tùy thuộc vào mức độ nghiêm trọng của bệnh và độ tuổi.
Vật lý trị liệu giúp cải thiện khả năng di chuyển, ngăn ngừa gãy xương và tăng sức mạnh cơ bắp thường hữu ích.
Điều trị gãy xương ở trẻ em như người lớn không bị viêm khớp. Một phương pháp điều trị chỉnh hình gọi là đặt thanh trong tủy (đặt các thanh vào xương) để định vị chân giúp hoạt động tốt hơn.
Một phương pháp điều trị mới hơn với thuốc biophosphonates đang được sử dụng để giúp hình thành xương và giảm nhu cầu phẫu thuật.
Dạng di truyền
Khi do đột biến gen COL1A1 hoặc COL1A2 gây ra, bệnh xương thủy tinh có kiểu di truyền trội trên nhiễm sắc thể thường, do đó chỉ cần một bản sao của gen bị thay đổi trong mỗi tế bào là đủ để gây bệnh. Nhiều người mắc bệnh loại I hoặc loại IV thừa hưởng đột biến từ cha hoặc mẹ mang bệnh. Hầu hết trẻ sơ sinh mắc các dạng khiếm khuyết nghiêm trọng hơn của quá trình tạo xương (như loại II và loại III) không có tiền sử bệnh trong gia đình. Ở những trẻ này, bệnh do đột biến mới (de novo) trong gen COL1A1 hoặc COL1A2. Loại V cũng được di truyền theo kiểu trội trên nhiễm sắc thể thường.

Nguồn: U.S. National Library of Medicine
Ít phổ biến hơn, bệnh xương thủy tinh có kiểu hình di truyền lặn trên nhiễm sắc thể thường, khi đó cần phải có cả hai bản sao đột biến của gen trong mỗi tế bào. Cha mẹ không bị bệnh, nhưng mỗi người mang một bản sao của gen bị đột biến. Các loại từ VI đến XVIII theo kiểu di truyền này.

Nguồn: U.S. National Library of Medicine
Xương thủy tinh loại XIX được di truyền theo kiểu gen lặn liên kết X, khi đó gen đột biến gây bệnh nằm trên nhiễm sắc thể X - một trong hai nhiễm sắc thể giới tính trong tế bào. Nam giới chỉ có một nhiễm sắc thể X, do đó đột biến tại bản sao gen duy nhất trong mỗi tế bào là đủ để gây ra bệnh. Nữ giới hai nhiễm sắc thể X, nên đột biến phải xảy ra ở cả hai bản sao của gen để gây bệnh. Bởi vì khó có khả năng nữ giới mang hai bản sao đột biến gen này, nam giới mang bệnh do đột biến gen lặn liên kết X thường xuyên hơn so với nữ. Một đặc điểm của di truyền liên kết X là người cha không thể truyền tính trạng liên kết X cho con trai.
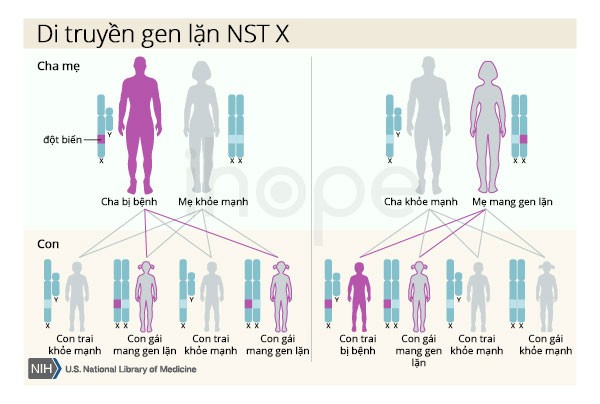
Nguồn: U.S. National Library of Medicine
Phòng ngừa
Hiện nay chưa có phương pháp phòng ngừa bệnh xương thủy tinh. Các cặp vợ chồng trước khi mang thai cần tư vấn di truyền nếu gia đình có tiền sử bệnh.
Nếu một người mắc bệnh xương thủy tinh loại I hay IV, khi sinh con sẽ có 50% khả năng di truyền bệnh cho con. Do đó, để bảo đảm 100% khả năng con không bị bệnh, cha mẹ có thể chọn phương pháp thụ tinh nhân tạo IVF và sàng lọc phôi PGS/PGD.
Trường hợp bệnh xương thủy tinh loại II hay III do đột biến mới (de novo) xảy ra trong quá trình tạo phôi, các cặp vợ chồng trong thời gian mang thai nên chủ động làm xét nghiệm sàng lọc thai NIPT ihope sớm ngay từ tuần thứ 9 để phát bệnh xương thủy tinh ở thai nhi.
Các tên gọi khác
- Bệnh xương giòn
- Fragilitas ossium
- OI
- Bệnh Vrolik
References
- Genetic Testing Information. Osteogenesis imperfecta. Retrieved May 27, 2022 from https://www.ncbi.nlm.nih.gov/gtr/conditions/C0029434/
- Catalog of Genes and Diseases from OMIM. OSTEOGENESIS IMPERFECTA, TYPE I; OI1. Retrieved May 27, 2022 from https://omim.org/entry/166200
- Genetic and Rare Diseases Information Center. Osteogenesis imperfecta. Retrieved May 27, 2022 from https://rarediseases.info.nih.gov/diseases/1017/osteogenesis-imperfecta
- U.S National Library of Medicine. Osteogenesis imperfecta. Retrieved May 27, 2022 from https://medlineplus.gov/osteogenesisimperfecta.html
- U.S National Library of Medicine. Osteogenesis imperfecta. Retrieved May 27, 2022 from https://medlineplus.gov/genetics/condition/osteogenesis-imperfecta/
- National Institute of Arthritis and Musculoskeletal and Skin Diseases. Osteogenesis imperfecta. Retrieved May 27, 2022 from https://www.niams.nih.gov/health-topics/osteogenesis-imperfecta
- National Human Genome Research Institute. Learning about Osteogenesis Imperfecta. Retrieved May 27, 2022 from https://www.genome.gov/Genetic-Disorders/Osteogenesis-Imperfecta
- National Institute of Arthritis and Musculoskeletal and Skin Diseases. What Is Osteogenesis Imperfecta? Retrieved May 27, 2022 from https://www.bones.nih.gov/health-info/bone/osteogenesis-imperfecta/overview
- Eunice Kennedy Shriver National Institute of Child Health and Human Development. What Are the Symptoms of Osteogenesis Imperfecta? Retrieved May 27, 2022 from https://www.nichd.nih.gov/health/topics/osteogenesisimp/conditioninfo/symptoms
- Eunice Kennedy Shriver National Institute of Child Health and Human Development. What are the treatments for osteogenesis imperfecta (OI)? Retrieved May 27, 2022 from https://www.nichd.nih.gov/health/topics/osteogenesisimp/conditioninfo/treatments
- Eunice Kennedy Shriver National Institute of Child Health and Human Development. How do health care providers diagnose osteogenesis imperfecta (OI)? Retrieved May 27, 2022 from https://www.nichd.nih.gov/health/topics/osteogenesisimp/conditioninfo/diagnose
- National Organization for Rare Disorders (NORD). Osteogenesis imperfecta. Retrieved May 27, 2022 from https://rarediseases.org/rare-diseases/osteogenesis-imperfecta/





