
Axit niệu 3-methylglutaconic (3-methylglutaconic aciduria – 3MGA) hay chứng thiếu men 3-methylglutaconyl-CoA hydratase là bệnh di truyền gây ra các vấn đề về thần kinh. Các bất thường có thể xuất hiện ngay sau sinh hoặc trong giai đoạn trẻ nhỏ.
Biểu hiện lâm sàng
Trẻ mắc axit niệu 3-methylglutaconic thường chậm phát triển các kỹ năng thể chất và vận động bao gồm:
- Chậm phát triển tâm thần vận động (psychomotor delay)
- Khó nói
- Rối loạn trương lực cơ dẫn đến co cơ không tự chủ (dystonia)
- Liệt cứng tứ chi
- Mất điều hòa vận động
Bệnh nhân có thể khởi phát các vấn đề mắt bao gồm teo thị giác. Bất thường này khiến tế bào thần kinh truyền thông tin từ mắt đến não bị teo. Khi bệnh tiến triển, người mắc axit niệu 3-methylglutaconic bị suy giảm thị lực nghiêm trọng.
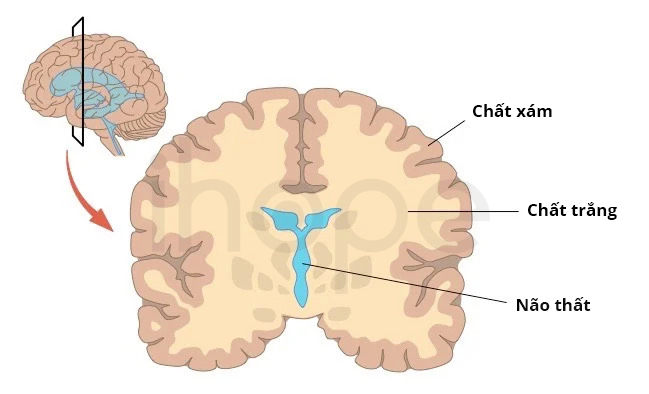
Trong một số trường hợp, các dấu hiệu và triệu chứng của bệnh axit niệu 3-methylglutaconic bắt đầu xuất hiện trong giai đoạn trưởng thành, chủ yếu từ 20 đến 30 tuổi. Phần lớn người bệnh bị bất thường chất trắng gọi là bệnh não chất trắng (leukoencephalopathy). Mô não này bao gồm các tế bào thần kinh có bao myelin, tế bào thân kinh đệm và tế bào hình sao. Chúng thực hiện nhiệm vụ liên lạc và truyền tín hiệu giữa các vùng não khác với nhau. Do đó, bất thường chất trắng dẫn đến các vấn đề thần kinh bao gồm rối loạn ngôn ngữ, mất điều hòa vận động, cứng khớp và mất trí nhớ.
Nguồn: Simply Psychology
Phần lớn những người biểu hiện các triệu chứng axit niệu 3-methylglutaconic trong giai đoạn trẻ nhỏ sẽ phát triển bệnh não chất trắng kèm theo bất thường thần kinh thị giác khi trưởng thành.
Cơ thể bệnh nhân tích tụ axit 3-methylglutaconic thừa đến mức gây ra chứng nhiễm toan chuyển hóa (máu thừa axit). Lượng chất này được bài tiết ra ngoài thông qua nước tiểu dẫn đến chứng axit niệu. Đây là biểu hiện quan trọng giúp bác sĩ chẩn đoán bệnh thông qua các xét nghiệm chuyên biệt bao gồm xét nghiệm nước tiểu và máu.
Độ phổ biến
Axit niệu 3-methylglutaconic là bệnh hiếm gặp, tỉ lệ mắc phải chưa được thống kê cụ thể. Khoảng 20 trường hợp đã được ghi nhận trên toàn thế giới.
Nguyên nhân
Đột biến gen AUH gây ra axit niệu 3-methylglutaconic. Gen AUH cung cấp hướng dẫn tạo ra enzym 3-methylglutaconyl-CoA hydratase. Enzyme này tham gia vào quá trình phân hủy axit amin leucine trong ti thể nhằm chuyển đổi năng lượng từ thực phẩm thành năng lượng tế bào sử dụng.
Đột biến gen AUH khiến enzyme 3-methylglutaconyl-CoA hydratase mất chức năng dẫn đến axit amin leucine không được xử lý đúng cách. Do đó, axit 3-methylglutaconic, axit 3-methylglutaric và axit 3-hydroxyisovaleric tích tụ trong cơ thể. Người ta cho rằng các chất này tích tụ trong dịch não tủy (chất lỏng bảo vệ xung quanh não và tủy sống) sẽ làm hỏng tế bào thần kinh, từ đó các dấu hiệu và triệu chứng bệnh khởi phát.
Độ tuổi khởi phát và mức độ tiến triển các triệu chứng khác nhau giữa mỗi bệnh nhân. Do đó, gen và yếu tố môi trường khác có thể ảnh hưởng đến biểu hiện của axit niệu 3-Methylglutaconic.
Chẩn đoán
Trước sinh
Chẩn đoán axit niệu 3-Methylglutaconic có thể được thực hiện trước sinh thông qua kiểm tra hàm lượng axit 3-hydroxyisovaleric cao trong nước ối. Phân tích enzyme từ tế bào ối nuôi cấy cũng có thể được thực hiện.
Sau sinh
Trong vòng 24–48 giờ sau khi sinh, trẻ được lấy máu gót chân để thực hiện sàng lọc sơ sinh nhằm phát hiện tình trạng axit niệu 3-methylglutaconic và các rối loạn khác.
Nếu kết quả sàng lọc sơ sinh cho thấy nguy cơ cao axit niệu 3-methylglutaconic, bác sĩ sẽ cho em bé làm thêm xét nghiệm để làm rõ. Điều quan trọng cần lưu ý là kết quả sàng lọc nguy cơ cao không đồng nghĩa với kết luận em bé mắc bệnh. Kết quả nguy cơ cao có thể do mẫu máu ban đầu được thu quá ít hoặc quá sớm. Tuy nhiên, cha mẹ nên nhớ đưa trẻ tái khám theo đúng lịch hẹn để làm xét nghiệm xác nhận. Nếu không được điều trị, thiếu men G6PD có thể ảnh hưởng sức khỏe của trẻ rõ rệt ngay sau khi sinh, xét nghiệm tiếp theo phải được tiến hành càng sớm càng tốt để xác định xem liệu trẻ có mắc bệnh hay không.
Xét nghiệm tiếp theo bao gồm kiểm tra mẫu nước tiểu và máu của bé nhằm đo lường nồng độ axit và chất độc có hại. Một số axit và độc tố tích tụ trong cơ thể khi trẻ mắc bệnh axit hữu cơ, vì vậy khảo sát nồng độ chất này trong cơ thể trẻ có thể giúp bác sĩ xác định xem trẻ có mắc bệnh hay không. Nồng độ acylcarnitine C5-OH cao trong máu và axit hữu cơ trong nước tiểu có thể cho biết trẻ mắc bệnh axit niệu 3-methylglutaconic. Đôi khi bác sĩ cần xét nghiệm thêm một mẫu da rất nhỏ.
Trường hợp khác
Bệnh axit niệu 3-methylglutaconic được chẩn đoán chủ yếu dựa trên các biểu hiện lâm sàng bao gồm bất thường vận động thể chất, teo thị giác, bệnh não chất trắng, nhiễm toan chuyển hóa và axit niệu. Tuy nhiên, những triệu chứng thường khác nhau giữa mỗi bệnh nhân và chúng không đặc hiệu cho bệnh. Do đó, bác sĩ thường chỉ định xét nghiệm hoạt tính 3-methylglutaconyl-CoA hydratase trong nguyên bào sợi hoặc bạch cầu, phân tích định lượng bài tiết axit hữu cơ qua nước tiểu nhằm nâng cao tính chính xác của chẩn đoán. Gần đây, phân tích dịch cơ thể bằng quang phổ NMR là phương pháp mới được sử dụng phổ biến. Ngoài ra, xét nghiệm di truyền phát hiện các đột biến gen AUH liên quan nhằm xác nhận các kết quả chẩn đoán.
Bệnh axit niệu 3-methylglutaconic cần được phân biệt giữa các loại (loại I, II và III) thông qua các chất chuyển hóa đặc biệt như axit 3-methylglutaconic.
Điều trị
Hiện nay, axit niệu 3-methylglutaconic chưa có phương pháp điều trị hoàn toàn. Liệu pháp ăn kiêng được sử dụng phổ biến. Trẻ mắc bệnh cần chế độ ăn hạn chế protein (chủ yếu là axit amin leucine) nhưng cần đảm bảo dinh dưỡng. Ngoài ra, một số trường hợp bổ sung L-carnitine có thể đem lại hiệu quả điều trị.
Dạng di truyền
Axit niệu 3-methylglutaconic di truyền theo kiểu lặn trên nhiễm sắc thể thường. Do đó, bệnh chỉ biểu hiện khi có cả hai bản sao của gen đột biến trong mỗi tế bào. Bệnh nhân mắc bệnh lặn trên nhiễm sắc thể thường sẽ có bố và mẹ mang một bản sao của gen đột biến, nhưng bố mẹ ít khi biểu hiện triệu chứng bệnh.

Nguồn: U.S. National Library of Medicine
Phòng ngừa
Bệnh axit niệu 3-methylglutaconic di truyền lặn do đột biến gen AUH, cha mẹ mang đột biến dị hợp nên gần như không có biểu hiện bệnh, do đó rất khó phát hiện cho đến khi sinh con. Để chủ động phòng ngừa, cha mẹ nên làm xét nghiệm sàng lọc gen lặn để chủ động cho tương lai của con. Các cặp vợ chồng trước khi mang thai cần tư vấn và xét nghiệm di truyền đảm bảo sinh con khỏe mạnh. Người thuộc nhóm nguy cơ mắc bệnh cần khám sức khỏe và tầm soát bệnh định kỳ.
Tên gọi khác
- 3-methylglutaconic aciduria, type I
- 3-MG-CoA-hydratase deficiency
- AUH defect
- MGA, type I
- MGA1
- MGCA1
- Primary 3-methylglutaconic aciduria
References
- Genetic Testing Information. 3-methylglutaconic aciduria type 1. Retrieved 25 September 2023 from https://www.ncbi.nlm.nih.gov/gtr/conditions/C0342727/
- Genetic and Rare Diseases Information Center. 3-methylglutaconic aciduria type 1. Retrieved 25 September 2023 from https://rarediseases.info.nih.gov/diseases/10321/3-methylglutaconyl-coa-hydratase-deficiency-auh-defect
- Catalog of Genes and Diseases from OMIM. 3-METHYLGLUTACONIC ACIDURIA, TYPE I; MGCA1. Retrieved 25 September 2023 from https://omim.org/entry/250950
- U.S National Library of Medicine. 3-methylglutaconyl-CoA hydratase deficiency. Retrieved 25 September 2023 from https://medlineplus.gov/genetics/condition/3-methylglutaconyl-coa-hydratase-deficiency/
- MalaCards. 3-Methylglutaconic Aciduria, Type I (MGCA1). Retrieved 25 September 2023 from https://www.malacards.org/card/3_methylglutaconic_aciduria_type_i_2
- National Organization for Rare Disorders. 3-methylglutaconyl-CoA hydratase deficiency (AUH defect). Retrieved 25 September 2023 from https://rarediseases.org/gard-rare-disease/3-methylglutaconyl-coa-hydratase-deficiency-auh-defect/
- Orphanet. 3-methylglutaconic aciduria type 1. Retrieved 25 September 2023 from https://www.orpha.net/consor/cgi-bin/OC_Exp.php?lng=EN&Expert=67046
- Baby's first test. 3-Methylglutaconic Aciduria. Retrieved 25 September 2023 from https://www.babysfirsttest.org/newborn-screening/conditions/3-methylglutaconic-aciduria
- National Library of Medicine. 3-Methylglutaconic Aciduria Type I Is Caused by Mutations in AUH. Retrieved 25 September 2023 from https://www.ncbi.nlm.nih.gov/pmc/articles/PMC378594/





