
Thiếu ACAD9 là bệnh gây ra các vấn đề về tim, yếu cơ và thiểu năng trí tuệ. Mức độ nghiêm trọng của bệnh biểu hiện khác nhau giữa từng người. Phần lớn bệnh nhân đều tích tụ axit lactic trong cơ thể. Ngoài ra, thiếu ACAD9 còn gây ra một số triệu chứng tại các cơ quan khác nhưng hiếm gặp.
Biểu hiện lâm sàng
Các dấu hiệu và triệu chứng khác nhau giữa 3 dạng bệnh:
- Người mắc chứng thiếu ACAD9 thể nhẹ thường cảm thấy buồn nôn và mệt mỏi khi thực hiện các hoạt động thể chất.
- Bệnh thể trung bình gây ra nhược cơ, chủ yếu các cơ vận động (cơ xương ).
- Bệnh thể nghiêm trọng phát triển các vấn đề về chức năng não kết hợp với bệnh cơ. Người bệnh có cơ tim phì đại, suy tim dẫn đến tử vong trẻ sơ sinh hoặc trẻ nhỏ.
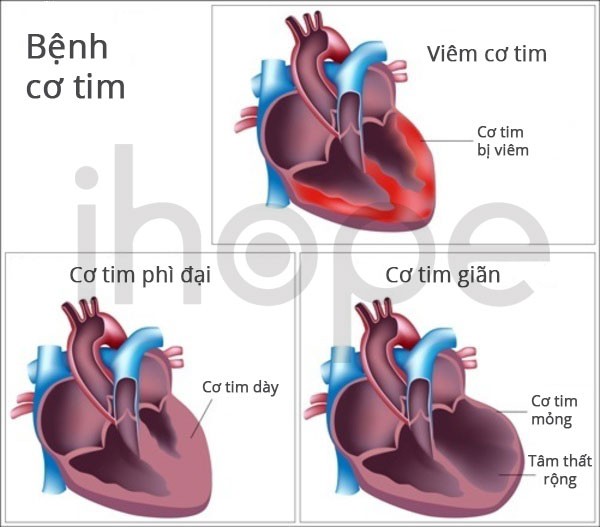
Nguồn: U.S. National Library of Medicine
Ảnh: Giải phẫu cơ
Nguồn: Medlineplus.gov
Trẻ mắc bệnh sống sót đến giai đoạn dậy thì thường bị thiểu năng trí tuệ kèm theo khởi phát các cơn co giật. Rối loạn vận động và các vấn đề về chức năng gan, thận là những biểu hiện hiếm gặp của bệnh.
Thiếu ACAD9 được phát hiện và điều trị kịp thời có thể giúp bệnh nhân cải thiện sức mạnh cơ bắp và giảm nồng độ axit lactic.
Độ phổ biến
Tỷ lệ mắc chứng thiếu ACAD9 chưa được thống kê cụ thể. Khoảng 25 trường hợp mắc bệnh đã được ghi nhận trên toàn thế giới.
Nguyên nhân
Đột biến gen ACAD9 gây ra chứng thiếu ACAD9. Gen ACAD9 cung cấp hướng dẫn tạo ra một enzyme có vai trò quan trọng trong quá trình lắp ráp nhóm protein phức hợp I. Phức hợp I tham gia phản ứng phosphoryl hóa oxy hóa tạo ra năng lượng trong ty thể.
Ngoài ra, enzyme ACAD9 trong ty thể còn thực hiện oxy hóa axit béo và chuyển hóa chúng thành năng lượng. Cụ thể hơn, enzyme ACAD9 chuyển hóa các axit béo chuỗi dài—nguồn năng lượng chính cho tim và cơ bắp. Trong giai đoạn nhịn ăn, axit béo giúp duy trì gan và các mô khác.
Một số đột biến gen ACAD9 làm mất hai chức năng quan trọng của enzyme dẫn đến các dấu hiệu và triệu chứng của bệnh. Từ đó bệnh nhân phát triển cơ tim phì đại, nhược cơ dù cơ chế vẫn chưa rõ ràng. Người ta cho rằng tế bào tạo ít năng lượng sẽ chết đi, chủ yếu các tế bào trong não, cơ và cơ quan khác. Số lượng tế bào ít dẫn đến khởi phát bệnh.
Chẩn đoán
Bệnh nhân bị nghi ngờ mắc chứng thiếu ACAD9 khi họ khởi phát các triệu chứng như nhược cơ, cơ tim phì đại, co giật và thiểu năng trí tuệ. Chẩn đoán bệnh dựa trên biểu hiện lâm sàng, tiền sử bệnh trong gia đình. Xét nghiệm di truyền nhằm phát hiện các đột biến gen liên quan là phương pháp chẩn đoán chính xác.
Điều trị
Hiện nay chưa có phương pháp điều trị dứt điểm chứng thiếu ACAD9. Các liệu pháp nhằm giảm nhẹ các triệu chứng, cải thiện chất lượng đời sống bệnh nhân.
Một số phương pháp có thể được chỉ định bao gồm:
- Ngăn hạ đường huyết
- Theo dõi thường xuyên chứng nhiễm toan axit lactic
- Bổ sung Vitamin B2 (riboflavin)
- Điều trị các bất thường về tim và gan
Trẻ có những biểu hiện như buồn ngủ quá mức, nôn mửa, tiêu chảy, sốt, chán ăn hoặc nhiễm trùng cần cấp cứu khẩn cấp.
Dạng di truyền
Thiếu ACAD9 di truyền theo kiểu lặn trên nhiễm sắc thể thường. Do đó, bệnh chỉ biểu hiện khi có cả hai bản sao của gen đột biến trong mỗi tế bào. Bệnh nhân mắc bệnh lặn trên nhiễm sắc thể thường sẽ có bố và mẹ mang một bản sao của gen đột biến, nhưng bố mẹ ít khi biểu hiện triệu chứng bệnh.

Nguồn: U.S. National Library of Medicine
Phòng ngừa
Bệnh di truyền lặn do đột biến gen ACAD9, cha mẹ mang đột biến dị hợp nên gần như không có biểu hiện bệnh, do đó rất khó phát hiện cho đến khi sinh con. Để chủ động phòng ngừa, cha mẹ nên làm xét nghiệm sàng lọc gen lặn để chủ động cho tương lai của con. Các cặp vợ chồng trước khi mang thai cần tư vấn và xét nghiệm di truyền đảm bảo sinh con khỏe mạnh. Người thuộc nhóm nguy cơ mắc bệnh cần khám sức khỏe và tầm soát bệnh định kỳ.
Các tên gọi khác
- Acyl-CoA dehydrogenase 9 deficiency
- Deficiency of acyl-CoA dehydrogenase family member 9
- Mitochondrial complex I deficiency due to ACAD9 deficiency
References
- Genetic Testing Information. Acyl-CoA dehydrogenase 9 deficiency. Retrieved 07 June 2023 from https://www.ncbi.nlm.nih.gov/gtr/conditions/C4747517/?_ga=2.208382032.374583242.1686122569-1635318420.1684225451
- Genetic and Rare Diseases Information Center. Mitochondrial complex I deficiency. Retrieved 07 June 2023 from https://rarediseases.info.nih.gov/diseases/3908/mitochondrial-complex-i-deficiency
- Catalog of Genes and Diseases from OMIM. MITOCHONDRIAL COMPLEX I DEFICIENCY, NUCLEAR TYPE 20; MC1DN20. Retrieved 07 June 2023 from https://omim.org/entry/611126
- U.S National Library of Medicine. ACAD9 deficiency. Retrieved 07 June 2023 from https://medlineplus.gov/genetics/condition/acad9-deficiency/
- Frontiers. Immunodeficiency with susceptibility to lymphoma with complex genotype affecting energy metabolism (FBP1, ACAD9) and vesicle trafficking (RAB27A). Retrieved 07 June 2023 from https://www.frontiersin.org/articles/10.3389/fimmu.2023.1151166/abstract
- MalaCards. Mitochondrial Complex I Deficiency, Nuclear Type 20 (MC1DN20). Retrieved 07 June 2023 from https://www.malacards.org/card/mitochondrial_complex_i_deficiency_nuclear_type_20
- National Organization for Rare Disorders. Very Long Chain Acyl CoA Dehydrogenase Deficiency (LCAD). Retrieved 07 June 2023 from https://rarediseases.org/rare-diseases/very-long-chain-acyl-coa-dehydrogenase-deficiency-lcad/
- Orphanet. Acyl-CoA dehydrogenase 9 deficiency. Retrieved 07 June 2023 from https://www.orpha.net/consor/cgi-bin/OC_Exp.php?lng=EN&Expert=99901
- Radiopaedia. Primary mitochondrial disorders. Retrieved 07 June 2023 from https://radiopaedia.org/articles/primary-mitochondrial-disorders




