
Hội chứng Gitelman hay hạ magie-hạ kali máu gia đình là nhóm bệnh di truyền gây mất cân bằng một số loại ion trong cơ thể như natri, kali, magie và canxi. Thận của người bệnh kém tái hấp thu muối vào trong máu, do đó nồng độ chất điện giải bị thay đổi. Các triệu chứng và mức độ nghiêm trọng của bệnh khác nhau giữa từng bệnh nhân, thậm chí ngay cả với thành viên trong cùng một gia đình. Hội chứng Gitelman không giống với các bệnh thận khác, triệu chứng bệnh thường khởi phát trong giai đoạn thiếu niên và trưởng thành.
Nguồn: U.S National Library of Medicine
Biểu hiện lâm sàng
Hội chứng Gitelman làm thay đổi nồng độ chất điện giải trong máu, từ đó một số triệu chứng phổ biến xuất hiện như:
- Khát nước
- Cơ co thắt không tự chủ
- Yếu cơ hoặc chuột rút
- Chóng mặt
- Buồn nôn
- Thèm thức ăn mặn
- Cảm giác châm chích hoặc ngứa ran trên da (dị cảm), đặc biệt là khuôn mặt
Ngoài ra, một số bệnh nhân có thể mắc các triệu chứng ít phổ biến hơn bao gồm:
- Hạ huyết áp
- Rối loạn nhịp tim
- Mệt mỏi có thể đi kèm kiệt sức
- Đau khớp do canxi hóa sụn
Phần lớn người bệnh biểu hiện triệu chứng nhẹ. Tuy nhiên, một số trường hợp bệnh nhân bị co rút cơ nghiêm trọng, tăng trưởng chậm, thậm chí tê liệt.
Độ phổ biến
Tỉ lệ mắc hội chứng Gitelman ước tính khoảng 1/40.000 người trên toàn thế giới.
Nguyên nhân
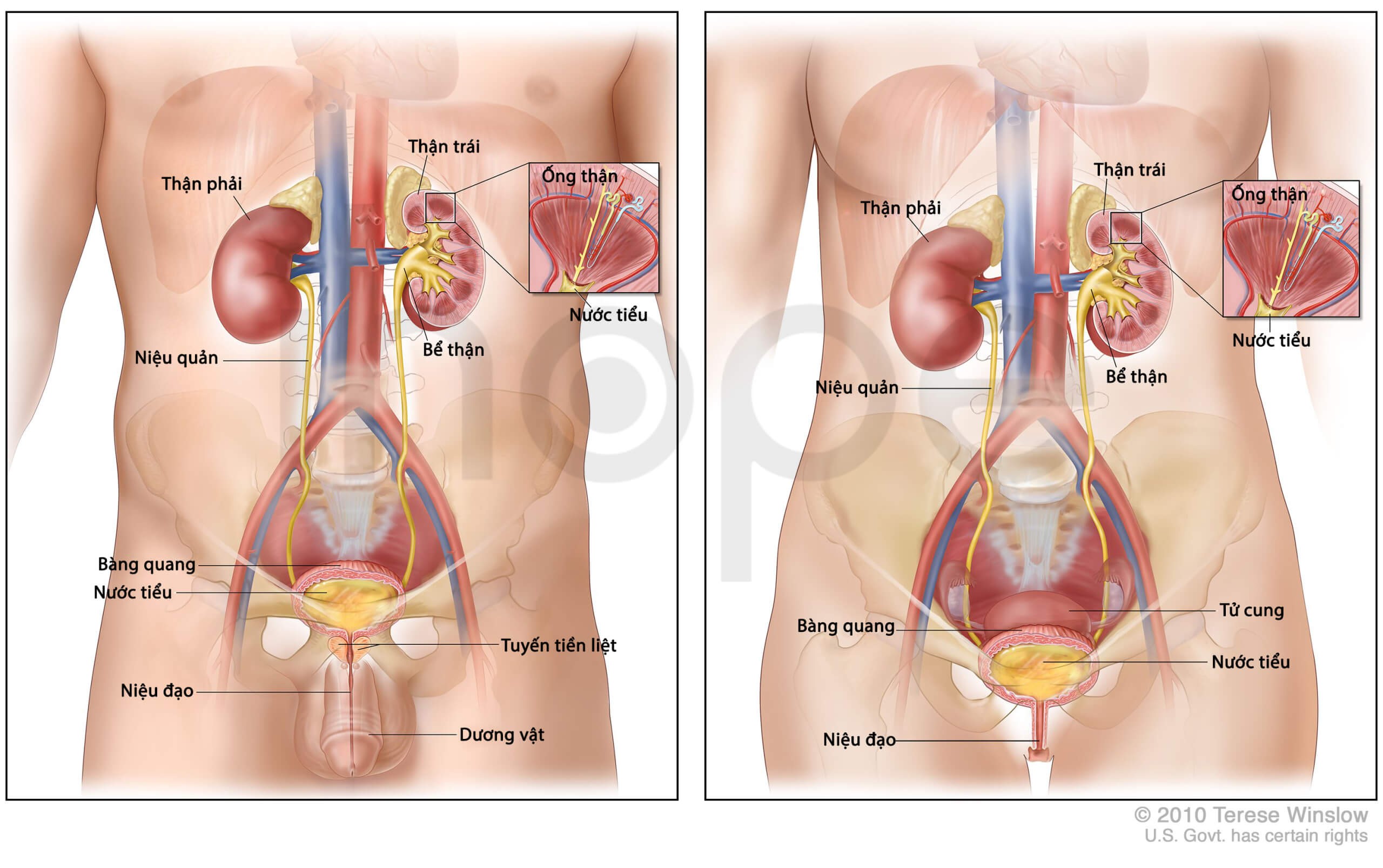
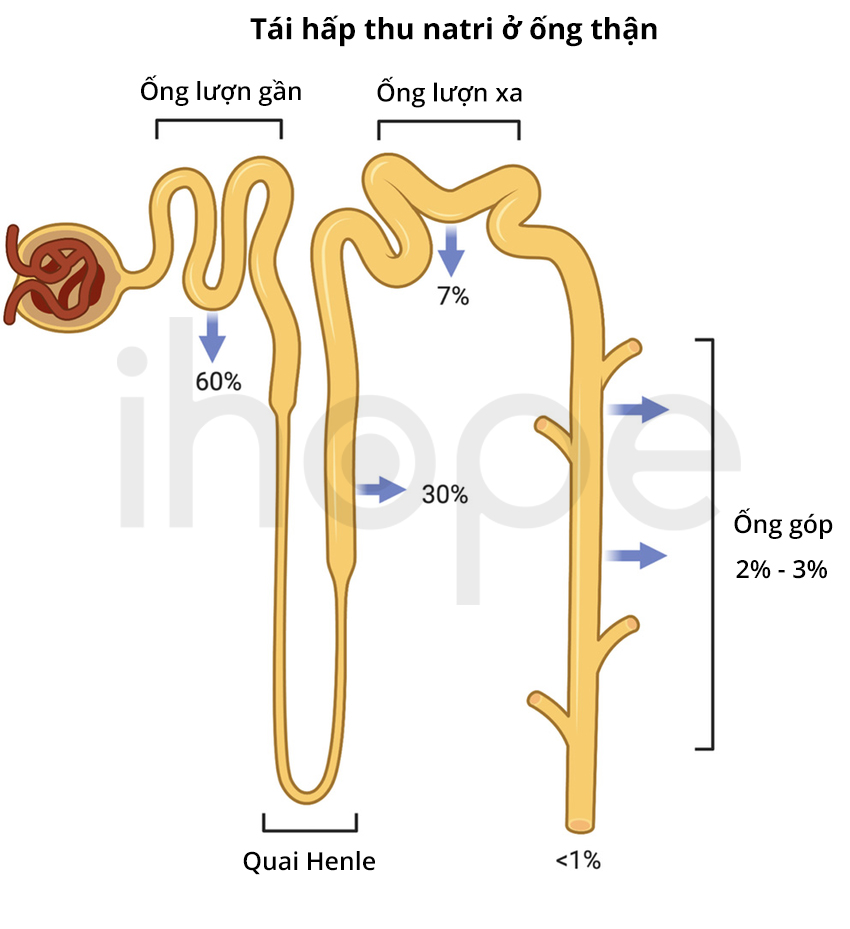
Đột biến gen SLC12A3 gây ra hội chứng Gitelman. Ngoài ra, người ta còn phát hiện đột biến gen CLCNKB có thể gây ra bệnh nhưng hiếm gặp. Gen SLC12A3 và SLC12A3 cung cấp hướng dẫn tạo ra protein tham gia cấu trúc nên kênh vận chuyển ion Na+. Các khoáng chất theo máu đến thận sẽ được cầu thận lọc vào khoang nước tiểu để tạo thành nước tiểu đầu tiên. Sau đó, nước tiểu tiếp tục di chuyển qua các ống thận từ ống lượn gần, quai Henle, ống lượn xa, ống góp rồi đổ vào bể thận. Trong quá trình này, nước và các ion cần thiết được tái hấp thu vào máu nhằm cân bằng chất điện giải trong cơ thể. Những chất không cần thiết được đào thải qua ngoài theo nước tiểu. Các kênh vận chuyển ion Na+ do gen SLC12A3 và SLC12A3 tạo ra hiện diện chủ yếu trong thận, chúng đảm nhiệm chức năng tái hấp thu muối (NaCl) từ nước tiểu vào trong máu.
Nguồn: ResearchGate
Phần lớn đột biến gen SLC12A3 và CLCNKB thay đổi axit amin đơn trong protein đồng vận chuyển NCC. Những đột biến này ngăn cản protein tiếp cận màng tế bào hoặc thay đổi khả năng vận chuyển ion natri và clorua. Các đột biến gen khác tạo ra một phiên bản protein ngắn bất thường và không có chức năng; chúng làm suy giảm khả năng tái hấp thu muối tại thận, dẫn đến cơ thể mất một lượng muối qua nước tiểu. Những bất thường trong protein NCC còn ảnh hưởng đến quá trình tái hấp thu các ion khác bao gồm kali, magie và canxi. Do đó, cơ thể mất cân bằng ion nên các triệu chứng của hội chứng Gitelman xuất hiện.
Chẩn đoán
Hội chứng Gitelman được chẩn đoán bằng nhiều phương pháp khác nhau. Ban đầu, bác sĩ xem xét tiền sử bệnh của cá nhân, gia đình và kiểm tra kỹ lưỡng các triệu chứng nhằm thu hẹp phạm vi chẩn đoán. Sau đó, bệnh nhân được kiểm tra thể chất nhằm đánh giá vấn đề chuột rút và yếu cơ. Ngoài ra, bác sĩ có thể thực hiện một số xét nghiệm chuyên biệt khác bao gồm:
- Xét nghiệm máu: giúp phát hiện tình trạng hạ kali máu, hạ magie máu, hạ clo huyết và tăng canxi máu
- Xét nghiệm renin và aldosterone: đánh giá nồng độ renin và aldosterone trong cơ thể
- Chẩn đoán phân biệt: loại trừ những bệnh có triệu chứng tương tự như hội chứng Bartter
- Xét nghiệm di truyền: kiểm tra đột biến gen SLC12A3 và SLC12A nhằm xác nhận kết quả chẩn đoán
Điều trị
Hội chứng Gitelman là bệnh di truyền nên hiện nay vẫn chưa có phương pháp điều trị dứt điểm bệnh. Phần lớn các liệu pháp đều hướng đến giảm nhẹ các triệu chứng nhằm cải thiện chất lượng đời sống bệnh nhân. Bác sĩ có thể chỉ định người bệnh dùng thuốc lợi tiểu kali (amiloride hoặc spironolactone) nhằm tăng nồng độ ion kali trong huyết thanh, hạn chế nhiễm toan và hạ magie máu. Ngoài ra, người bệnh có thể chủ động bổ sung thêm kali, natri clorua và magie theo hướng dẫn của bác sĩ.
Dạng di truyền
Hội chứng Gitelman di truyền theo kiểu lặn trên nhiễm sắc thể thường. Do đó, bệnh chỉ biểu hiện khi có cả hai bản sao của gen đột biến trong mỗi tế bào. Bệnh nhân mắc bệnh lặn trên nhiễm sắc thể thường sẽ có bố và mẹ mang một bản sao của gen đột biến, nhưng bố mẹ ít khi biểu hiện triệu chứng bệnh.

Nguồn: U.S. National Library of Medicine
Phòng ngừa
Hội chứng Gitelman di truyền lặn, cha mẹ mang đột biến dị hợp gần như không có biểu hiện bệnh, do đó rất khó phát hiện cho đến khi sinh con. Để chủ động phòng ngừa, cha mẹ nên làm xét nghiệm sàng lọc gen lặn để chủ động cho tương lai của con. Các cặp vợ chồng trước khi mang thai cần tư vấn và xét nghiệm di truyền nhằm đảm bảo sinh con khỏe mạnh. Người thuộc nhóm nguy cơ mắc bệnh cần khám sức khỏe và tầm soát bệnh định kỳ.
Các tên gọi khác
- Familial hypokalemia-hypomagnesemia
- Gitelman's syndrome
- GS
- Hypokalemia-hypomagnesemia, primary renotubular, with hypocalciuria
- Tubular hypomagnesemia-hypokalemia with hypocalcuria
References
- Genetic Testing Information. Familial hypokalemia-hypomagnesemia. Retrieved January 12, 2024 from https://www.ncbi.nlm.nih.gov/gtr/conditions/C0268450/
- Genetic and Rare Diseases Information Center. Gitelman syndrome. Retrieved January 12, 2024 from https://rarediseases.info.nih.gov/diseases/8547/gitelman-syndrome
- Catalog of Genes and Diseases from OMIM. GITELMAN SYNDROME; GTLMNS. Retrieved January 12, 2024 from https://omim.org/entry/263800
- MedlinePlus. Gitelman syndrome. Retrieved January 12, 2024 from https://medlineplus.gov/genetics/condition/gitelman-syndrome/
- Osmosis. Gitelman Syndrome. Retrieved January 12, 2024 from https://www.osmosis.org/answers/Gitelman-syndrome





