Loạn sản sụn chấm liên-kết-X dạng 2 (X-linked chondrodysplasia punctata 2) là hội chứng di truyền hiếm gặp, bắt nguồn từ đột biến gen EBP trên nhiễm sắc thể X. Phần lớn bệnh nhân là nữ. Một số biểu hiện đặc trưng của hội chứng bao gồm da có vảy, rụng tóc theo từng mảng, tầm vóc thấp, cơ thể bất đối xứng và khuôn mặt dị biệt.
Biểu hiện lâm sàng
Bệnh lí về xương
Trẻ mắc hội chứng có cấu trúc xương bất thường. Hình ảnh chụp X-quang cho thấy nhiều đốm nhỏ xuất hiện tại các đầu xương hoặc sụn. Triệu chứng này thường xuất hiện đối với trẻ sơ sinh, mất đi khi trẻ lớn lên, ảnh hưởng đến quá trình phát triển của xương dài tại tay và chân, xương sườn, sụn khí quản.
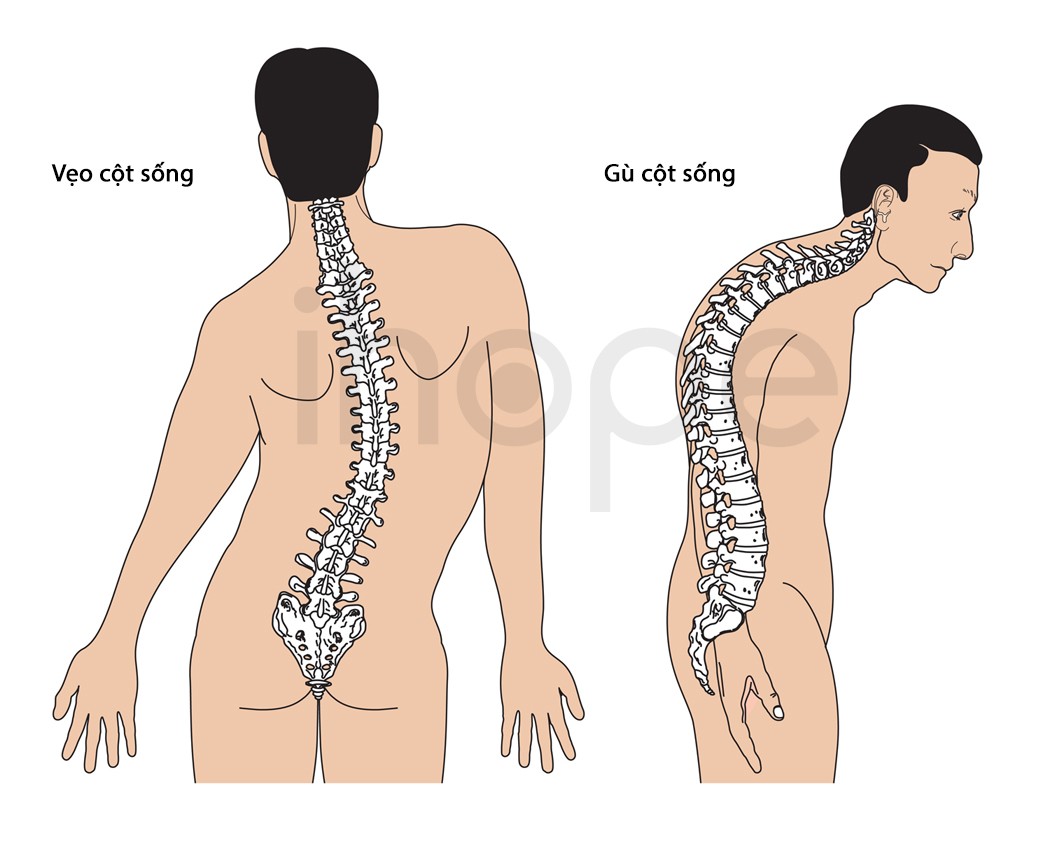
Hội chứng có thể dẫn đến các dị tật xương như:
- Xương cánh tay ngắn
- Xương đùi ngắn
- Vẹo cột sống
- Gù lưng
- Chiều dài tay, chân không đều
- Nhiều ngón
- Co cứng khớp
- Bàn chân khoèo
- Trật khớp hông
- Trật khớp xương bánh chè
- Tầm vóc thấp bé
Nguồn: Blamb/Shutterstock.com
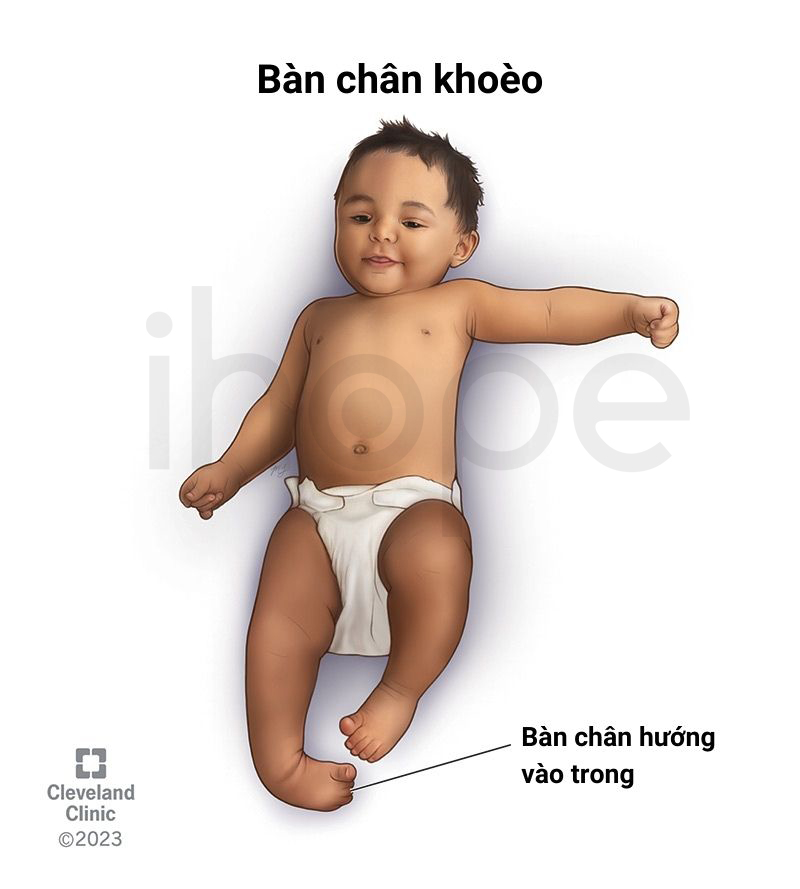
Nguồn: Cleveland Clinic
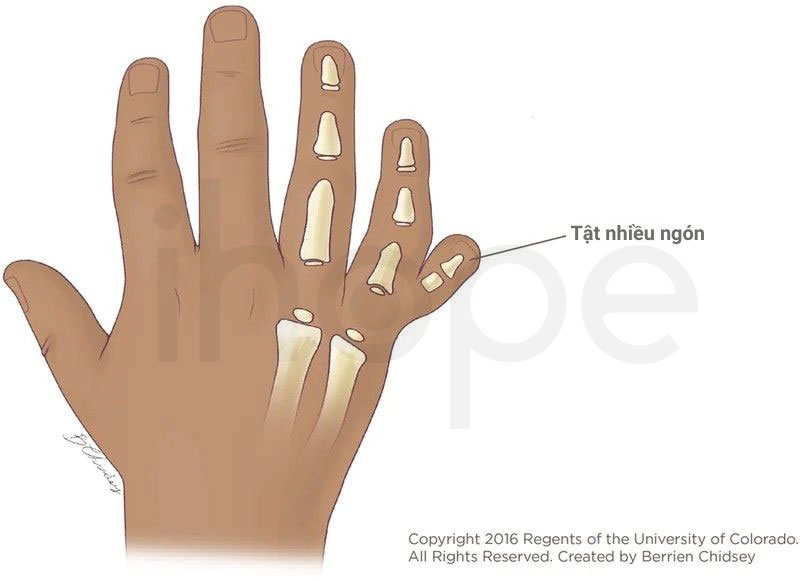
Nguồn: Children's Hospital Colorado
Khuôn mặt dị biệt
Trẻ mắc hội chứng loạn sản sụn chấm liên-kết-X dạng 2 thường có các đặc điểm khuôn mặt như:
- Sống mũi lõm
- Lông mày, lông mi thưa
- Sọ lồi
- Tai thấp
- Khe mi mắt hướng xuống
- Vòm miệng cao
- Khoảng cách giữa hai mắt xa nhau
Bệnh lí về mắt
Trẻ gặp nhiều vấn đề về mắt như:
- Đục thủy tinh thể
- Mắt nhỏ
- Giác mạc nhỏ
- Giảm thị lực
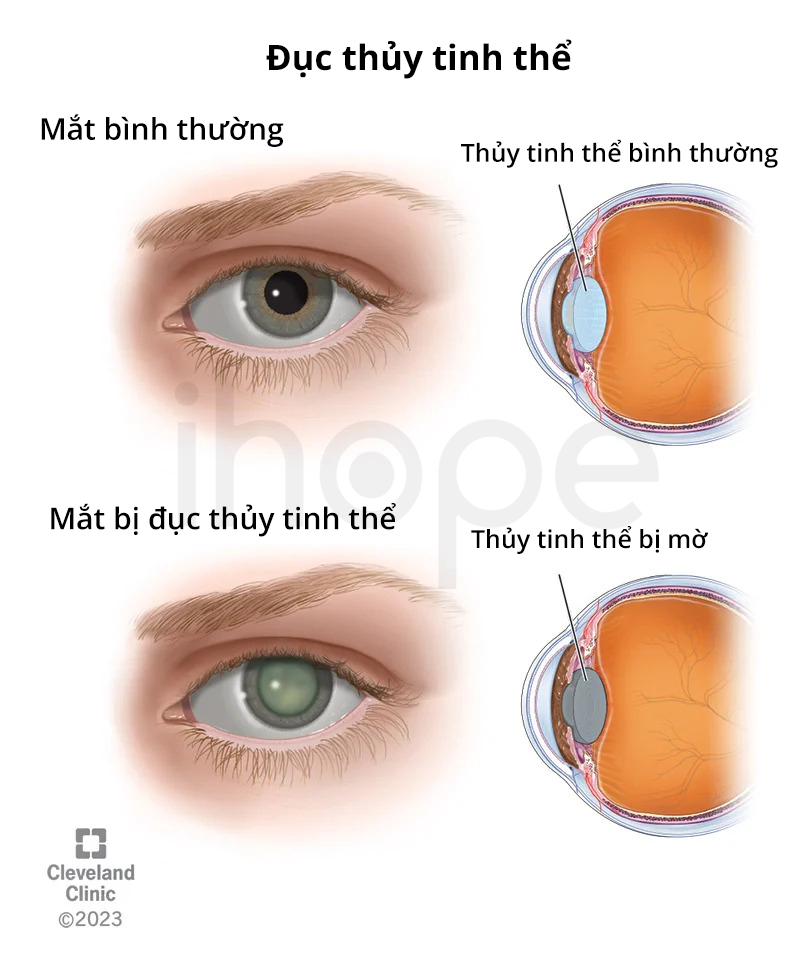
Nguồn: Cleveland Clinic
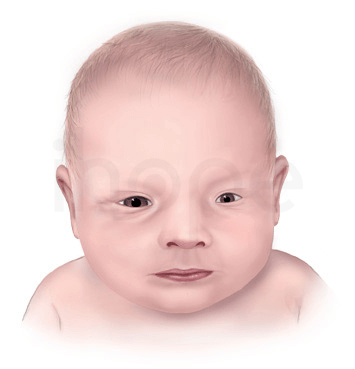
Nguồn: Centers for Disease Control and Prevention
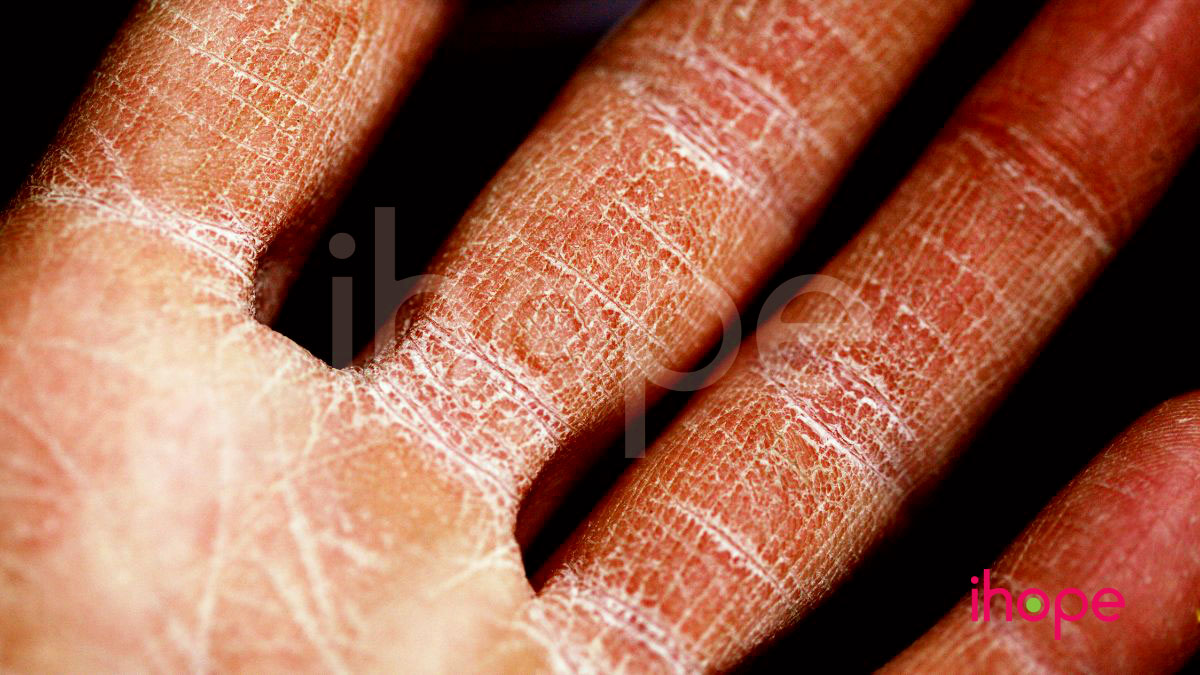
Bệnh lí về da và tóc
Bệnh ảnh hưởng đến quá trình phát triển của da và tóc, dẫn đến một số bất thường như:
- Da khô có vảy
- Tăng sắc tố da
- Đỏ da toàn thân
- Teo nang lông
- Tóc rụng thành từng mảng
- Tóc thô, không bóng
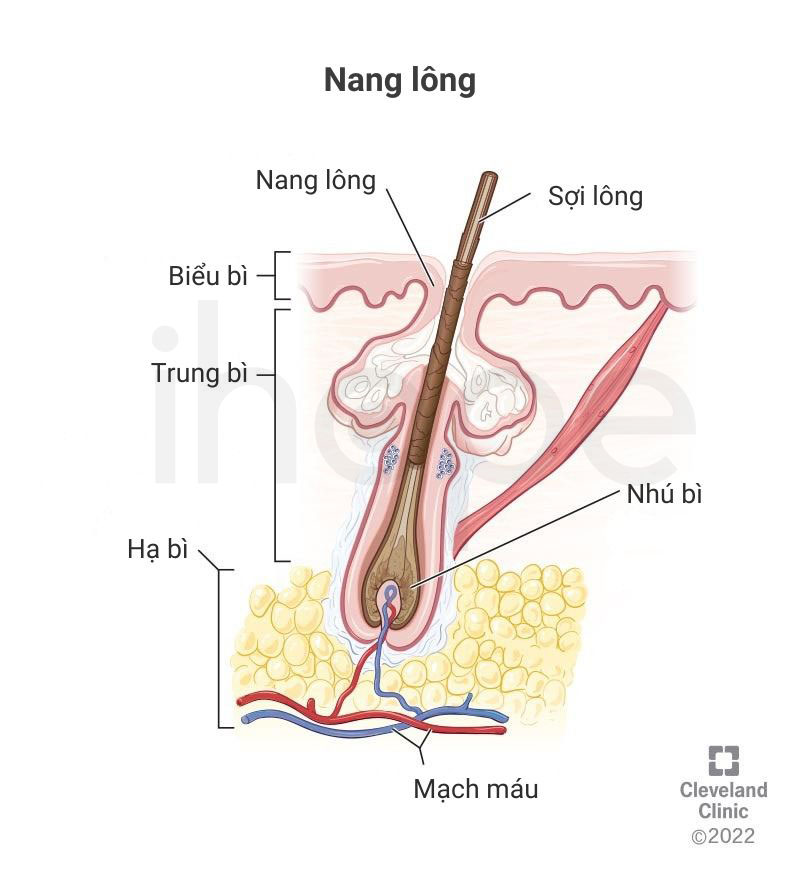
Nguồn: Cleveland Clinic
Triệu chứng khác
Ngoài ra, bệnh nhi có một số triệu chứng khác như:
- Thận ứ nước
- Hạ huyết áp
- Loạn sản vành tai
- Giảm thính lực
- Móng phẳng
- Móng tách rời
- U nang màng nhện
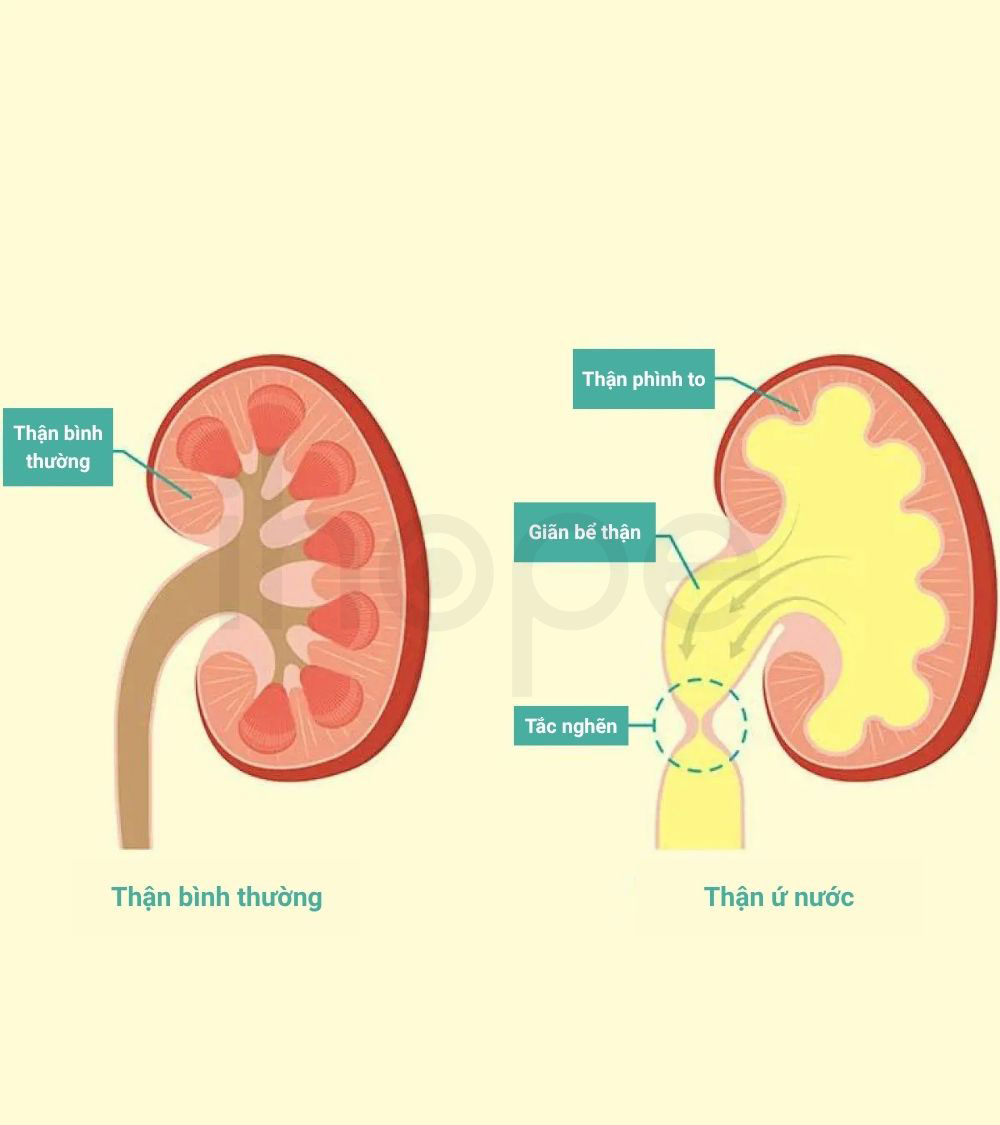
Nguồn: MomJunction
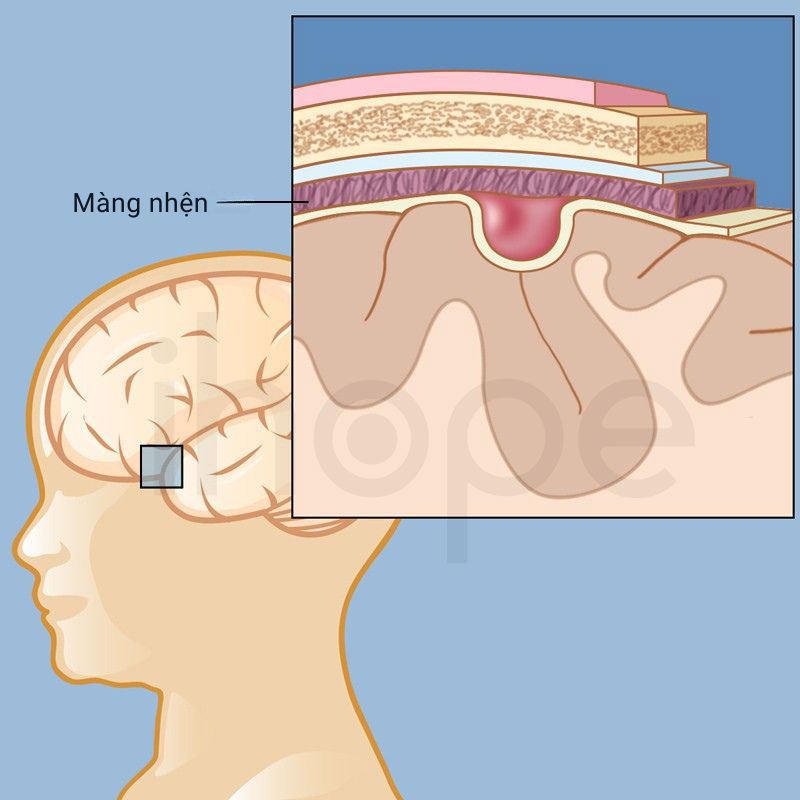
Nguồn: Weill Cornell Medicine
Độ phổ biến
Phần lớn người bệnh là nữ, trong khi bệnh nhân nam thường tử vong từ khi còn trong bụng mẹ. Người ta ghi nhận khoảng 10 nam giới mắc hội chứng này. Ước tính tỉ lệ mắc bệnh trên toàn thế giới khoảng 1/100.000–200.000 người.
Nguyên nhân
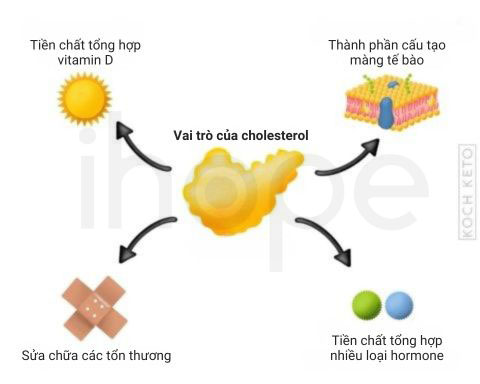
Hội chứng loạn sản sụn chấm liên-kết-X dạng 2 do đột biến gen EBP gây ra. Gen EBP cung cấp hướng dẫn tạo enzyme 3β-hydroxysteroid-Δ8,Δ7-isomerase. Enzyme này tham gia sản xuất cholesterol—phân tử quan trọng đối với quá trình phát triển bình thường của phôi thai. Sau khi trẻ được sinh ra, cholesterol tiếp tục tham gia vào các hoạt động phát triển hệ thần kinh, ổn định cấu trúc tế bào và tổng hợp hormone.
Đột biến gen EBP làm gián đoạn quá trình sản xuất 3β-hydroxysteroid-Δ8,Δ7-isomerase, dẫn đến thiếu hụt cholesterol. Đồng thời, do cholesterol không được tổng hợp, các tiền chất của cholesterol tích tụ trong cơ thể gây độc tế bào. Khi đó, lượng cholesterol thấp kết hợp với các chất trung gian từ con đường tổng hợp cholesterol tích tụ gây ảnh hưởng nghiêm trọng đến quá trình tăng trưởng và phát triển của trẻ.
Đột biến thường gây bất hoạt một phần gen đối với bệnh nhân nữ do hiện tượng bất hoạt X không hoàn toàn, trong khi đối với bệnh nhân nam, đột biến thường gây tử vong sớm trong thai kỳ.
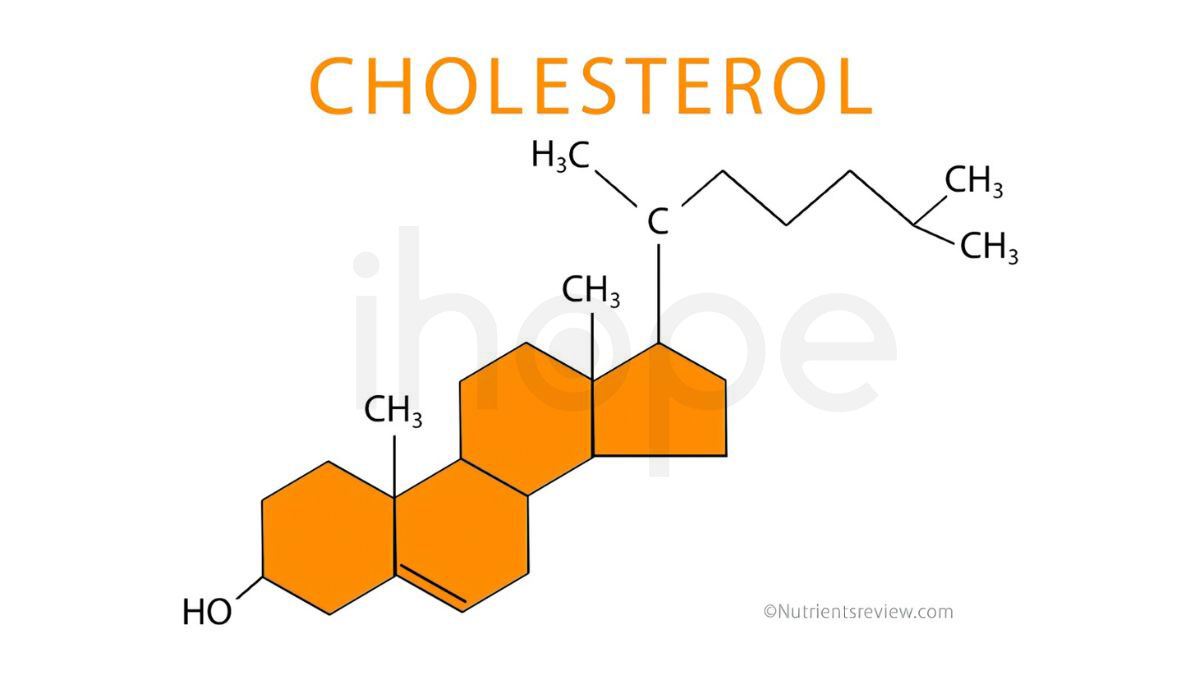
Nguồn: NutrientsReview
Nguồn: Koch Keto
Chẩn đoán
Hội chứng loạn sản sụn chấm liên-kết-X dạng 2 được chẩn đoán dựa trên biểu hiện lâm sàng kết hợp với kết quả xét nghiệm sinh hoá, hình ảnh, sinh thiết và di truyền.
Xét nghiệm sinh hoá
Bác sĩ tiến hành kiểm tra nồng độ sterol trong máu nhằm đưa ra chẩn đoán ban đầu. Bệnh nhân mắc hội chứng loạn sản sụn chấm liên kết thể X dạng 2 có nồng độ sterol trong máu cao bất thường.
Chẩn đoán hình ảnh
Đồng thời, kết quả chụp X-quang xương giúp bác sĩ phát hiện những đốm nhỏ trong cấu trúc xương của trẻ khi còn trong bụng mẹ.
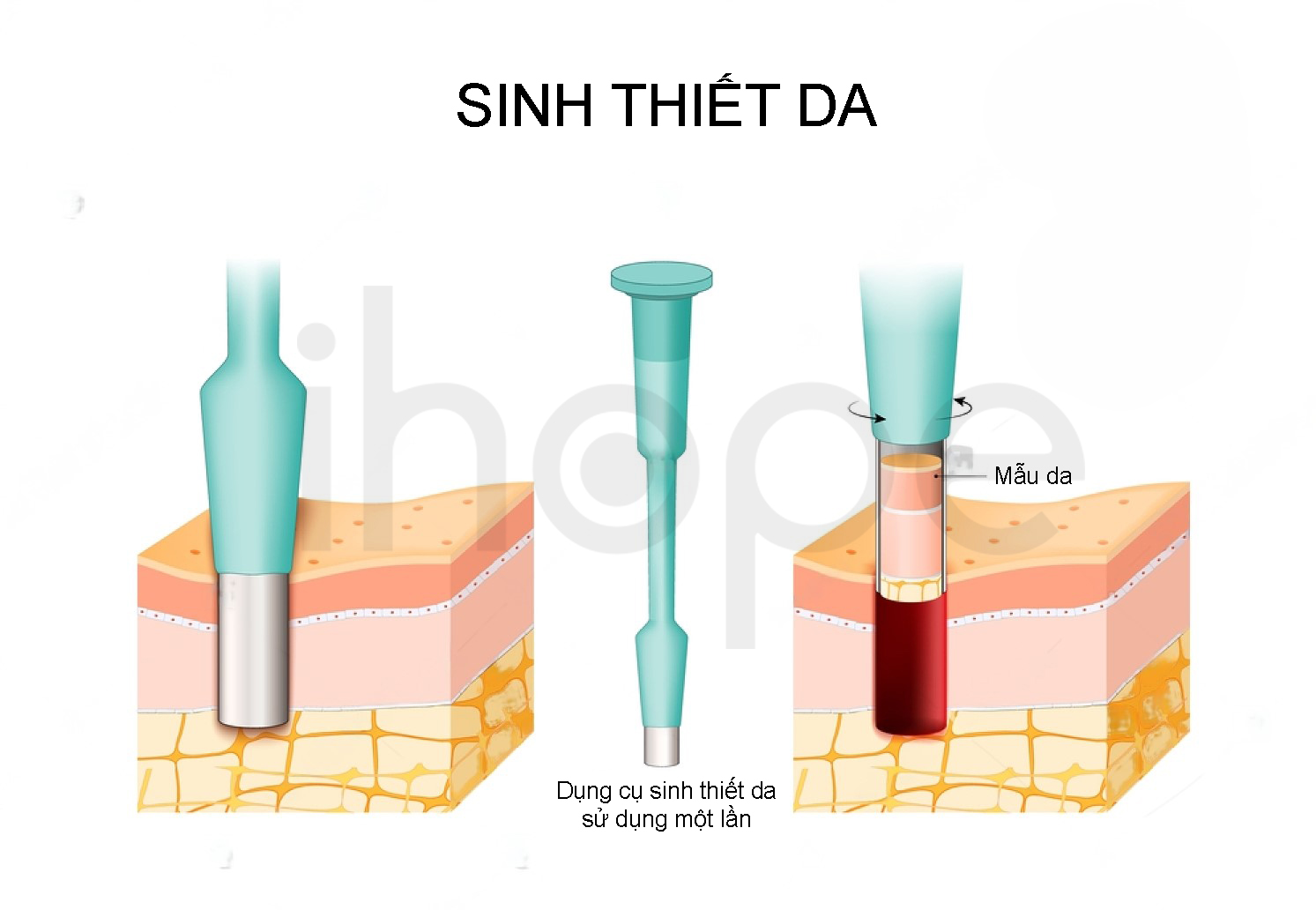
Sinh thiết
Ngoài ra, bác sĩ có thể sinh thiết da của bệnh nhân nhằm đánh giá triệu chứng sừng hóa lỗ chân lông và lắng đọng canxi keratin.
Nguồn: Skin and Hair Academy
Xét nghiệm di truyền
Kết quả chẩn đoán có thể được xác nhận bằng những xét nghiệm di truyền nhằm phát hiện đột biến gen EBP.
Điều trị
Hiện nay, chưa có phương pháp điều trị hoàn toàn hội chứng loạn sản sụn chấm liên-kết-X dạng 2. Các liệu pháp chủ yếu nhằm giảm nhẹ triệu chứng và cải thiện chất lượng đời sống người bệnh.
Một số liệu pháp hỗ trợ bao gồm:
- Phẫu thuật điều trị dị tật
- Vật lí trị liệu
- Hạn chế tiếp xúc với ánh nắng mặt trời
Dạng di truyền
Hội chứng loạn sản sụn chấm liên-kết-X dạng 2 di truyền theo kiểu trội trên nhiễm sắc thể giới tính X. Nam giới chỉ có một nhiễm sắc thể X, do đó một bản sao của gen đột biến trong mỗi tế bào đủ gây ra triệu chứng bệnh nghiêm trọng. Người cha bị bệnh không thể truyền gen đột biến này cho con trai. Đối với phụ nữ có hai nhiễm sắc thể X, đột biến tại một bản sao của gen thường chỉ gây ra các triệu chứng nhẹ.

Nguồn: U.S. National Library of Medicine
Phòng ngừa
Hội chứng loạn sản sụn chấm liên-kết-X dạng 2 có cơ chế di truyền liên kết X phức tạp nên khó phát hiện trên những người phụ nữ mang gen bệnh cho đến khi sinh con. Người mẹ nên làm xét nghiệm sàng lọc gen lặn để chủ động cho tương lai của con. Các cặp vợ chồng trước khi mang thai cần tư vấn và xét nghiệm di truyền nhằm đảm bảo sinh con khỏe mạnh. Người thuộc nhóm nguy cơ mắc bệnh cần khám sức khỏe và tầm soát bệnh định kỳ.
Các tên gọi khác
- CDPX2
- Chondrodysplasia punctata 2, X-linked dominant
- Chondrodysplasia punctata caused by mutation in EBP
- EBP chondrodysplasia punctata
- Chondrodysplasia punctata 2, X-linked
- Conradi-Hünermann syndrome
- Conradi-Hünermann-Happle syndrome
- Happle syndrome
- X-linked dominant chondrodysplasia punctata
References
- MedlinePlus. X-linked chondrodysplasia punctata 2. Retrieved June 22, 2025 from https://medlineplus.gov/genetics/condition/x-linked-chondrodysplasia-punctata-2/
- Genetic and Rare Diseases Information Center. X-linked chondrodysplasia punctata 2. Retrieved June 22, 2025 from https://rarediseases.info.nih.gov/diseases/6189/x-linked-chondrodysplasia-punctata-2
- GeneReviews. Chondrodysplasia Punctata 2, X-Linked. Retrieved June 22, 2025 from https://www.ncbi.nlm.nih.gov/sites/books/NBK55062/
- ONIM. CHONDRODYSPLASIA PUNCTATA 2, X-LINKED DOMINANT; CDPX2. Retrieved June 22, 2025 from https://www.omim.org/entry/302960
- Orphanet. X-linked dominant chondrodysplasia punctata. Retrieved June 22, 2025 from https://www.orpha.net/en/disease/detail/35173