Loạn sản khớp hông và vai dạng chấm (rhizomelic chondrodysplasia punctata) ảnh hưởng đến quá trình phát triển nhiều bộ phận trên cơ thể. Biểu hiện đặc trưng của người bệnh bao gồm bất thường xương, khuôn mặt dị biệt, thiểu năng trí tuệ và các vấn đề hô hấp.
Hiện nay, người ta đã xác định ba loại loạn sản khớp hông và vai dạng chấm gồm loại 1 (RCDP1), loại 2 (RCDP2) và loại 3 (RCDP3). Chúng có biểu hiện lâm sàng tương tự nhau và khác nhau về nguyên nhân di truyền.
Biểu hiện lâm sàng
Bất thường xương
Loạn sản khớp hông và vai dạng chấm khiến người bệnh co rút xương cánh tay, xương đùi, dị tật khớp làm khớp cứng và đau. Bên cạnh đó, bệnh nhân có biểu hiện loạn sản sụn dạng chấm, chúng ảnh hưởng đến quá trình phát triển của xương dài và có thể chẩn đoán thông qua chụp X-quang.
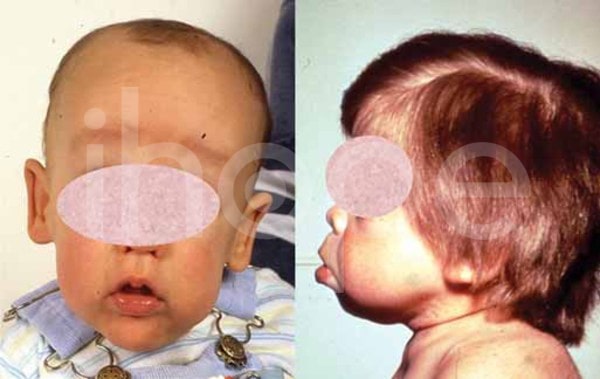
Khuôn mặt dị biệt
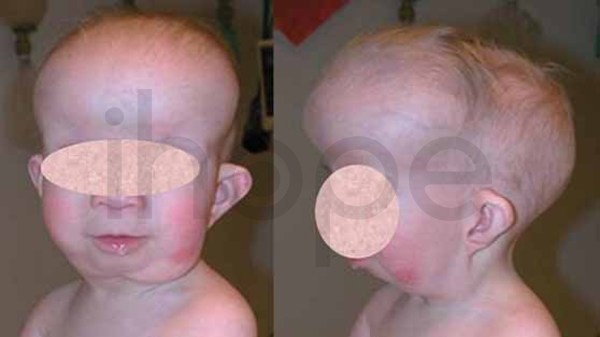
Ngoài ra, người bệnh có khuôn mặt dị biệt với những điểm đặc trưng sau:
- Trán gồ
- Hai mắt cách xa nhau
- Phần giữa mặt trũng xuống (giảm sản giữa mặt )
- Mũi nhỏ với lỗ mũi hếch
- Má phồng to
Phần lớn bệnh nhân đều bị đục thủy tinh thể, triệu chứng này xuất hiện ngay khi trẻ sinh ra hoặc khởi phát trong giai đoạn sơ sinh.
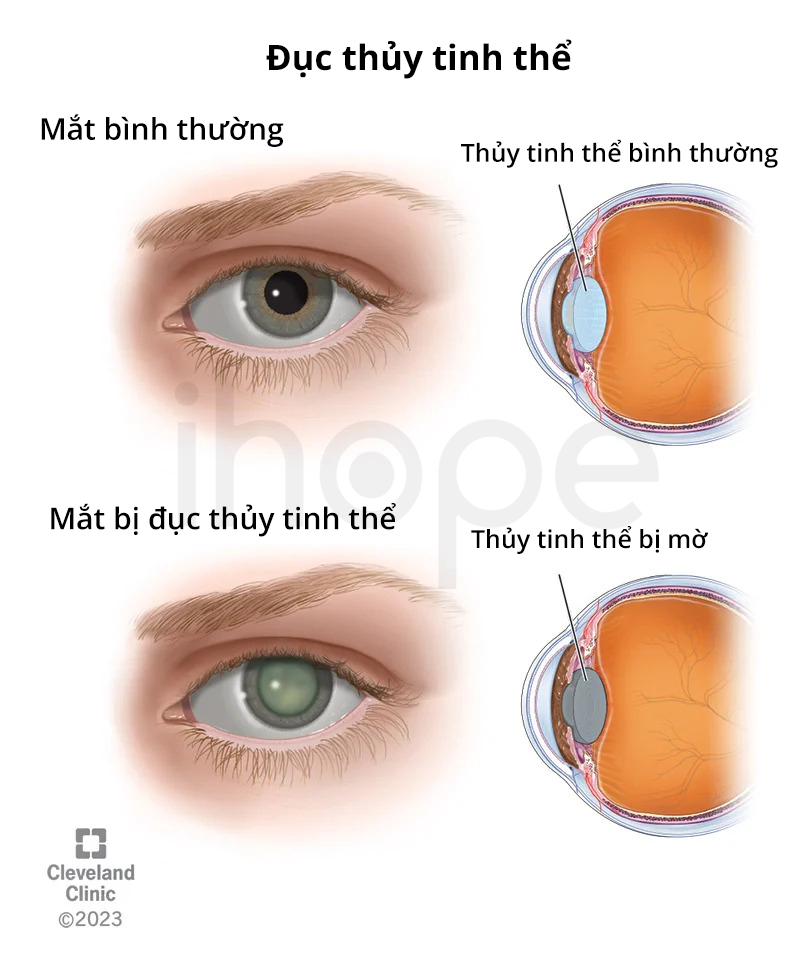
Nguồn: Cleveland Clinic
Ảnh: Trán nhô ra phía trước
Nguồn: Elements of Morphology, National Human Genome Research Institute
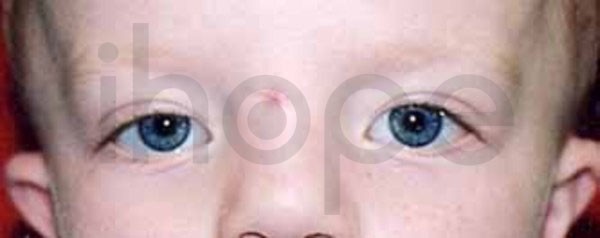
Ảnh: Khoảng cách giữa hai mắt rộng
Nguồn: Elements of Morphology, National Human Genome Research Institute
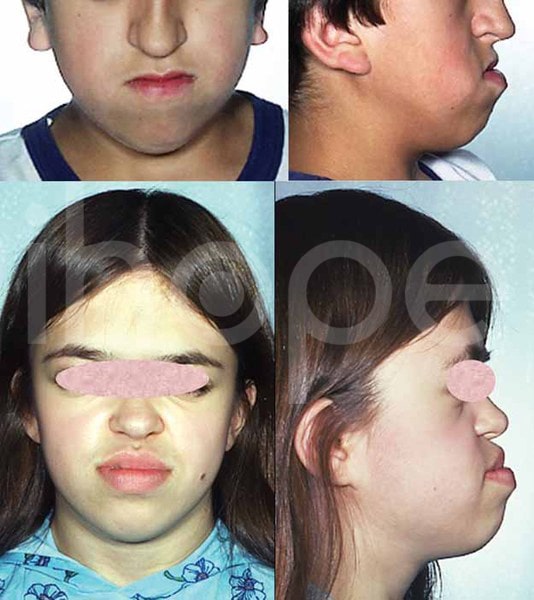
Ảnh: giảm sản vùng giữa mặt
Nguồn: Elements of Morphology, National Human Genome Research Institute
Ảnh: Má đầy đặn
Nguồn: Elements of Morphology, National Human Genome Research Institute
Thiểu năng trí tuệ
Người bệnh chậm phát triển và thiểu năng trí tuệ nghiêm trọng. Đa số người bệnh không đạt các mốc phát triển bình thường như tự ngồi không cần hỗ trợ, khả năng cầm, nắm hoặc diễn đạt một câu. Trẻ sơ sinh mắc bệnh tăng trưởng chậm so với những trẻ khác cùng tuổi và thường bị co giật.
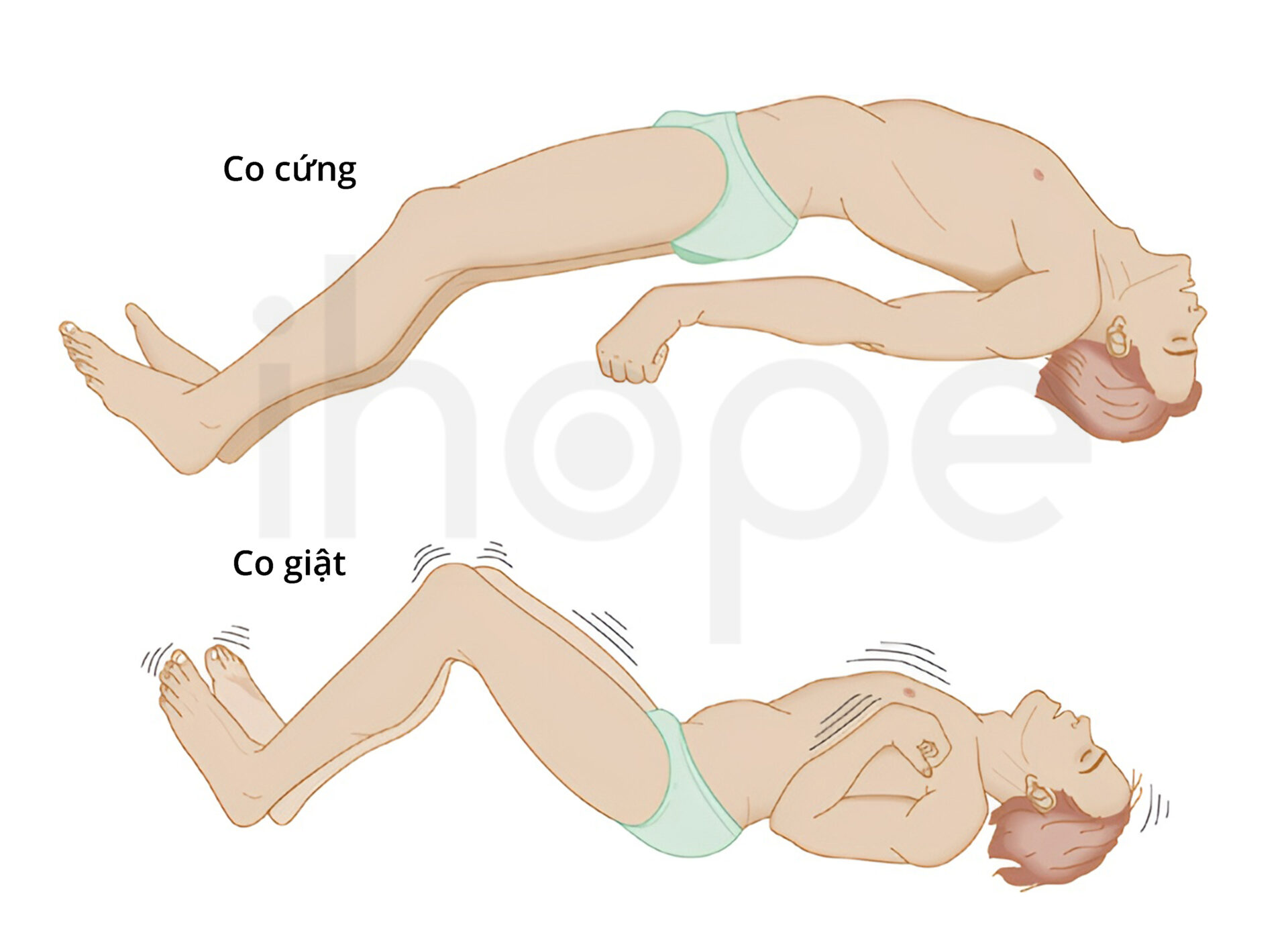
Nguồn: NEUROSURGERY Neurosurgery.
Vấn đề về hô hấp
Bên cạnh đó, người bệnh thường xuyên nhiễm trùng đường hô hấp tái phát và mắc các bệnh liên quan đến hô hấp gây đe dọa tính mạng. Do loạn sản khớp hông và vai dạng chấm dẫn đến nhiều vấn đề sức khỏe nghiêm trọng, phần lớn người bệnh không thể sống quá 10 tuổi. Tuy nhiên, một vài cá nhân biểu hiện bệnh nhẹ hơn có thể sống đến đầu trưởng thành.
Nguồn: National Cancer Institute.
Độ phổ biến
Người ta ước tính tỉ lệ mắc loạn sản khớp hông và vai dạng chấm là 1/100.000 người trên toàn thế giới. Loạn sản khớp hông và vai dạng chấm loại 1 phổ biến hơn hai loại còn lại.
Nguyên nhân
Đột biến gen PEX7, GNPAT hoặc AGPS gây ra loạn sản khớp hông và vai dạng chấm. Trong đó, đột biến gen PEX7 phổ biến nhất, chúng là nguyên nhân gây ra bệnh loại 1. Đột biến gen GNPAT, AGPS lần lượt gây ra loạn sản khớp hông và vai dạng chấm loại 2 và 3.
Ba gen này liên quan đến quá trình hình thành và hoạt động chức năng của bào quan peroxisome. Peroxisome hiện diện trong tế bào nhân thực, chúng chứa các enzyme cần thiết nhằm phân hủy axit béo và một số hợp chất độc hại. Bên cạnh đó, peroxisome tham gia sản xuất những loại lipid để sử dụng trong quá trình tiêu hóa và đảm bảo chức năng của hệ thần kinh, ví dụ như lipid plasmalogen.
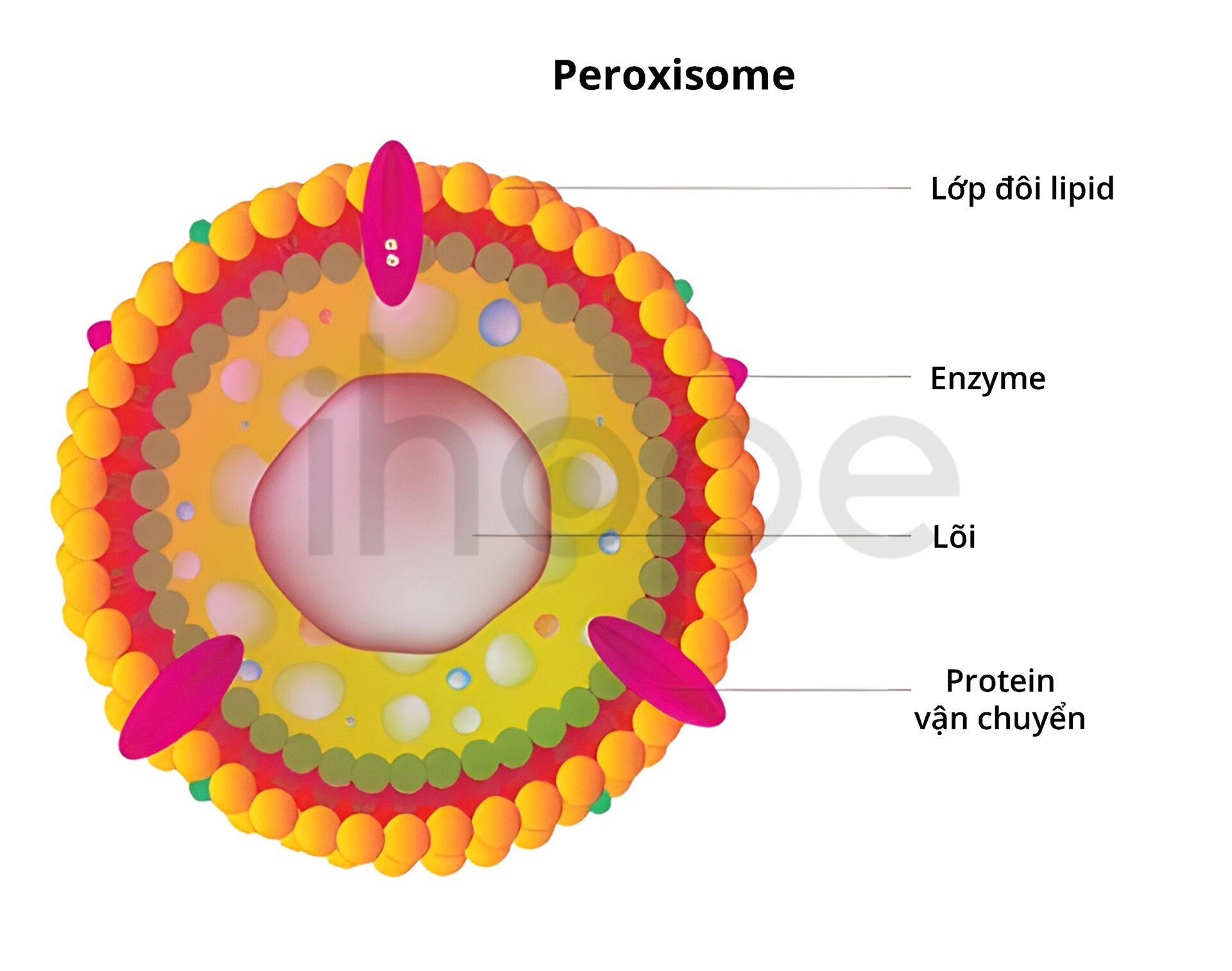
Nguồn: Shutterstock.
Trong peroxisome, ba gen này cung cấp hướng dẫn tạo ra các protein tham gia quá trình tổng hợp phân tử lipid plasmalogen. Plasmalogen hiện diện trong màng tế bào khắp cơ thể, tuy nhiên, người ta chưa biết nhiều về chức năng của phân tử này.
Đột biến gen PEX7, GNPAT hoặc AGPS làm gián đoạn quá trình peroxisome tổng hợp ra plasmalogen. Hiện nay, người ta đang tìm hiểu mối liên hệ giữa quá trình tổng hợp plasmalogen và các biểu hiện lâm sàng của bệnh loạn sản khớp hông và vai dạng chấm.
Chẩn đoán
Loạn sản khớp hông và vai dạng chấm được chẩn đoán dựa trên biểu hiện lâm sàng. Trẻ sơ sinh nghi ngờ mắc bệnh có các biểu hiện như đục thủy tinh thể, khuôn mặt dị biệt, xương cánh tay và xương đùi ngắn. Bên cạnh đó, bác sĩ có thể chỉ định người bệnh thực hiện xét nghiệm sinh hóa và di truyền phân tử nhằm xác nhận kết quả chẩn đoán.
Ngoài ra, bác sĩ có thể dự đoán thời điểm người bệnh có dấu hiệu co giật bằng cách đo điện não đồ (EEG) và quan sát hình ảnh não. Phương pháp này giúp phát hiện hoạt động điện bất thường và suy giảm chất trắng trong não, do đó, bác sĩ đánh giá quá trình tiến triển bệnh và đưa ra biện pháp can thiệp kịp thời.

Nguồn: verywellhealth.com
Điều trị
Phương pháp điều trị loạn sản khớp hông và vai dạng chấm được cá nhân hóa dựa trên độ tuổi, phân loại và mức độ biểu hiện của người bệnh. Biện pháp can thiệp chủ yếu bao gồm phẫu thuật và vật lí trị liệu. Hiện nay chưa có phương pháp điều trị bệnh hoàn toàn, do đó, quá trình điều trị tập trung kiểm soát các triệu chứng cụ thể và nâng cao chất lượng đời sống cho bệnh nhân.
Một số liệu pháp điều trị phổ biến bao gồm:
- Đặt ống dẫn thức ăn
- Theo dõi chức năng phổi và vật lí trị liệu hô hấp
- Phẫu thuật đục thủy tinh thể nhằm cải thiện thị lực
- Vật lí trị liệu và phẫu thuật chỉnh hình nhằm điều trị co cứng biến dạng khớp
- Đo lường mức plasmalogen trong cơ thể
Đối với bệnh nhân thuộc loại 1, họ cần duy trì chế độ ăn ít acid phytanic nhằm ngăn ngừa acid phytanic tích tụ và làm tổn thương tế bào. Bên cạnh đó, người bệnh cần đo lường mức acid phytanic định kì hằng năm nhằm theo dõi mức độ bệnh tiến triển.
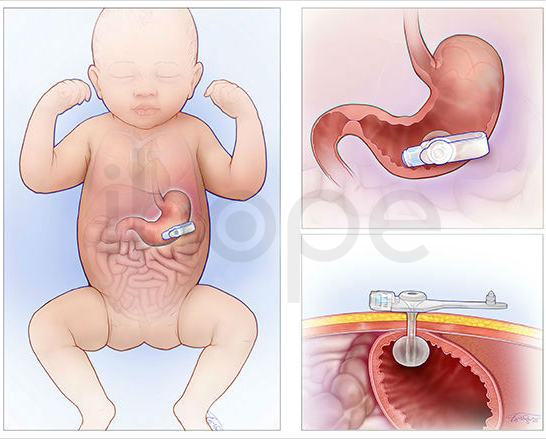
Ảnh: Phẫu thuật mở dạ dày để đặt ống truyền thức ăn
Nguồn: Children's Hospital of Phialadenphia
Dạng di truyền
Loạn sản khớp hông và vai dạng chấm di truyền theo kiểu lặn trên nhiễm sắc thể thường. Do đó, bệnh chỉ biểu hiện khi có cả hai bản sao của gen đột biến trong mỗi tế bào. Bệnh nhân mắc bệnh lặn trên nhiễm sắc thể thường sẽ có bố và mẹ mang một bản sao của gen đột biến, nhưng họ ít khi biểu hiện triệu chứng bệnh.

Nguồn: U.S. National Library of Medicine
Phòng ngừa
Loạn sản khớp hông và vai dạng chấm di truyền lặn do đột biến nhiều gen, cha mẹ mang đột biến dị hợp nên gần như không có biểu hiện bệnh, do đó rất khó phát hiện cho đến khi sinh con. Cha mẹ nên làm xét nghiệm sàng lọc gen lặn để chủ động cho tương lai của con. Các cặp vợ chồng trước khi mang thai cần tư vấn và xét nghiệm di truyền nhằm đảm bảo sinh con khỏe mạnh. Người thuộc nhóm nguy cơ mắc bệnh cần khám sức khỏe và tầm soát bệnh định kì.
Các tên gọi khác
- RCP
- RCDP
- Chondrodysplasia punctata, rhizomelic
References
- Genetic Testing Information. Rhizomelic chondrodysplasia punctata type 1. Retrieved August 20, 2024 from https://www.ncbi.nlm.nih.gov/gtr/conditions/C1859133/
- Genetic and Rare Diseases Information Center. Rhizomelic chondrodysplasia punctata. Retrieved August 20, 2024 from https://rarediseases.info.nih.gov/diseases/13160/index
- Catalog of Genes and Diseases from OMIM. RHIZOMELIC CHONDRODYSPLASIA PUNCTATA, TYPE 1; RCDP1. Retrieved August 20, 2024 from https://omim.org/entry/215100
- U.S National Library of Medicine. Rhizomelic chondrodysplasia punctata. Retrieved August 20, 2024 from https://medlineplus.gov/genetics/condition/rhizomelic-chondrodysplasia-punctata/
- MalaCards. Rhizomelic Chondrodysplasia Punctata (RCP). Retrieved August 20, 2024 from https://www.malacards.org/card/rhizomelic_chondrodysplasia_punctata
- National Institute of Health. The neurology of rhizomelic chondrodysplasia punctata. Retrieved August 20, 2024 from https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4228450/
- Orphanet. Rhizomelic chondrodysplasia punctata. Retrieved August 20, 2024 from https://www.orpha.net/en/disease/detail/177
- Radiopaedia. Chondrodysplasia punctata. Retrieved August 20, 2024 from https://radiopaedia.org/articles/chondrodysplasia-punctata