
Loạn sản xương hàm mặt kèm tật đầu nhỏ (Mandibulofacial dysostosis with microcephaly) là bệnh di truyền gây ra các bất thường tại đầu và mặt. Bệnh nhân thường có kích thước đầu nhỏ bất thường tại thời điểm sinh và đầu không phát triển cùng tốc độ với cơ thể, dẫn đến tật đầu nhỏ tiến triển.
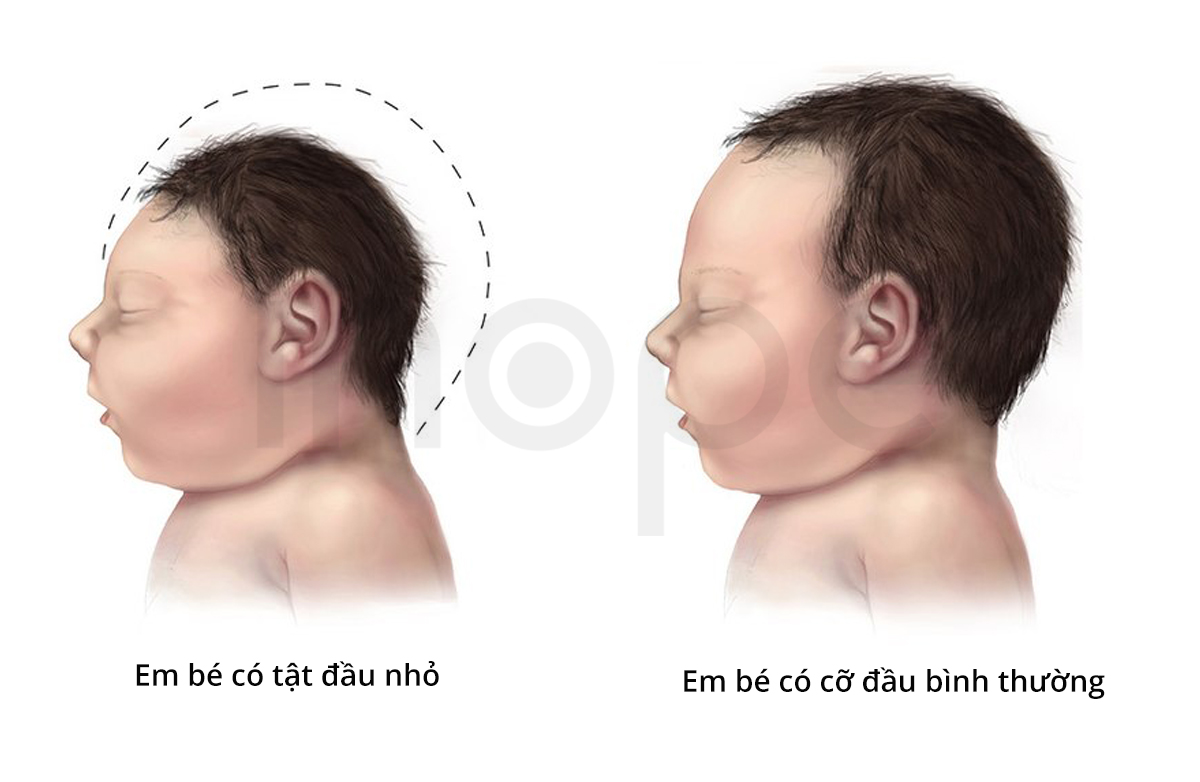
Ảnh: Tật đầu nhỏ
Nguồn: Centers for Disease Control and Prevention
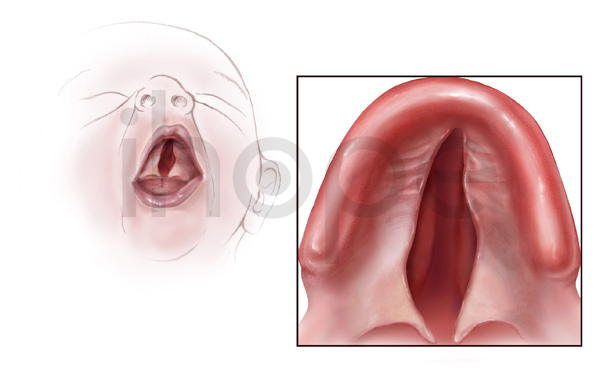
Ảnh: Hở hàm ếch
Nguồn: Health Centers for Disease Control and Prevention
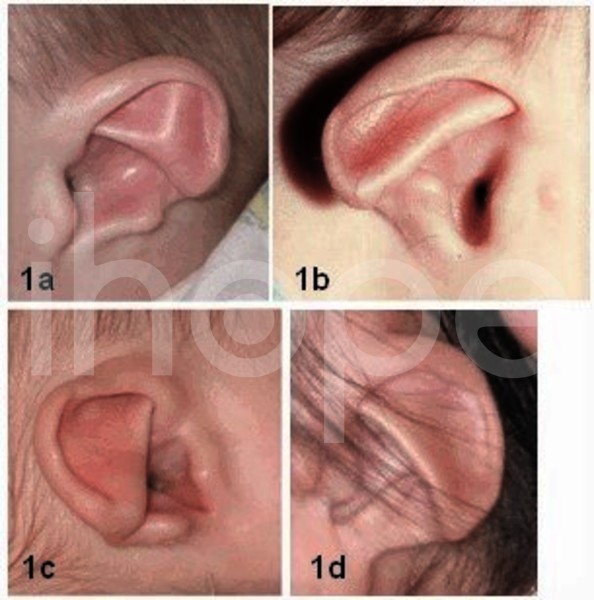
Ảnh: Dị dạng tai ngoài
Ảnh 1a) Tai cụp và không có thùy. Ảnh 1b) phần giữa tai kém phát triển và không có thùy. Ảnh 1c) tai cụp với phần giữa tai quá phát triển và không có thùy. Ảnh 1d) tai mỏng với phần giữa của tai nổi rõ.
Nguồn: GeneReviews, University of Washington
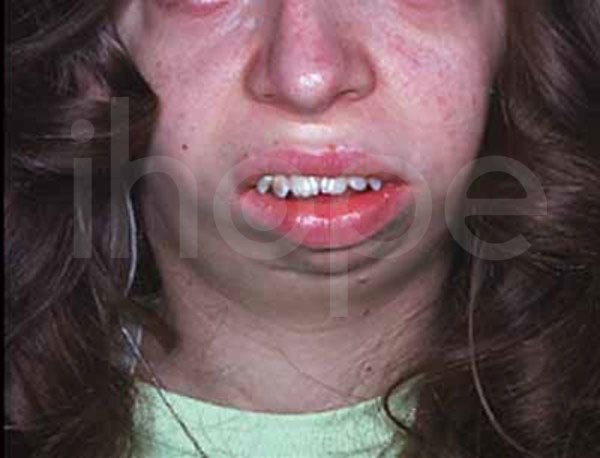
Ảnh: Xương hàm và xương cằm nhỏ, thu hẹp
Nguồn: U.S. National Library of Medicine
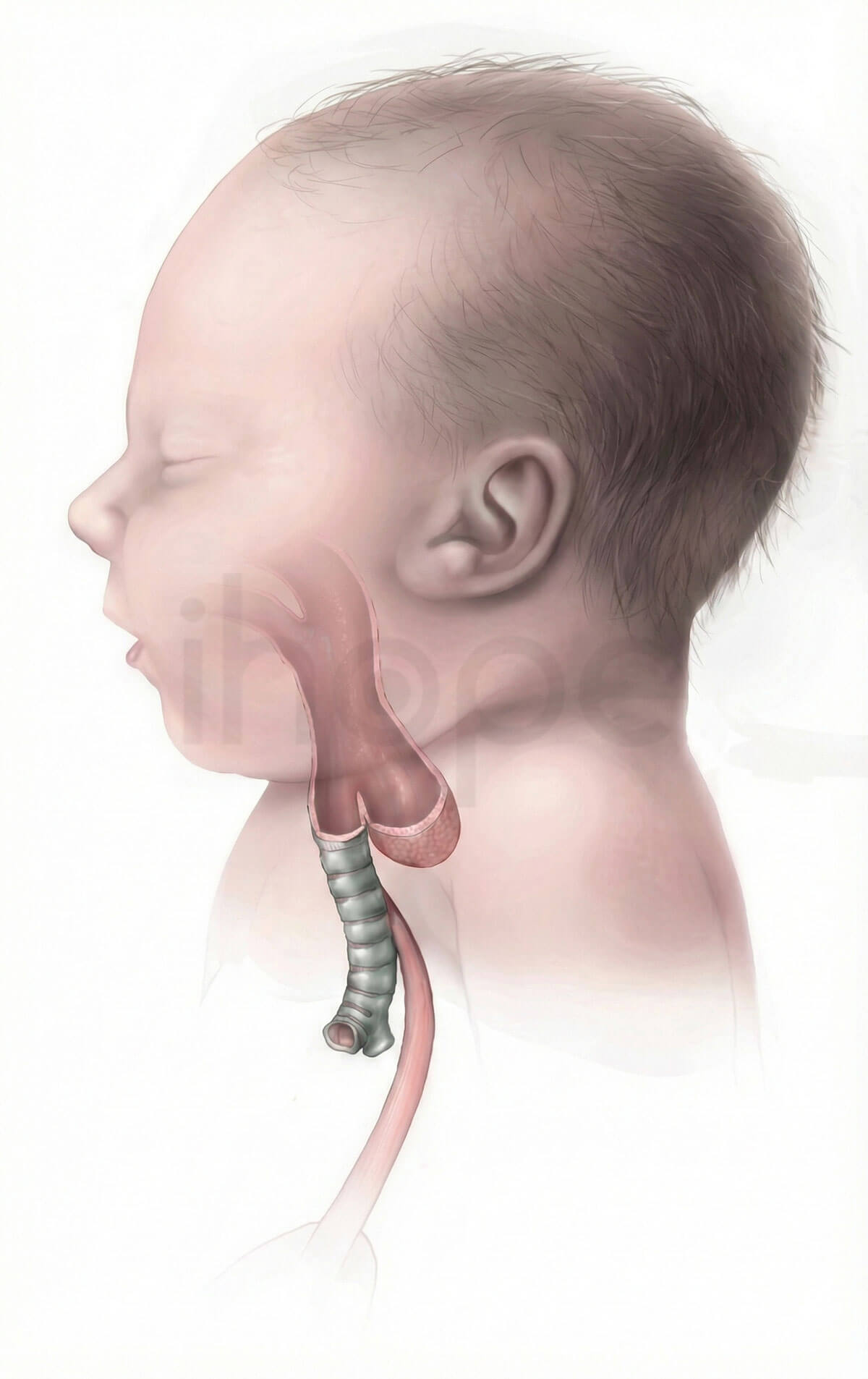
Ảnh: Teo thực quản
Nguồn: Centers for Disease Control and Prevention[/caption]
Biểu hiện lâm sàng
Người mắc loạn sản xương hàm mặt và tật đầu nhỏ có biểu hiện chậm phát triển và thiểu năng trí tuệ từ mức độ nhẹ đến nặng, kèm theo các vấn đề ngôn ngữ.
Các đặc điểm dị biệt trên khuôn mặt bao gồm:
- Thiểu sản vùng giữa mặt và xương gò má
- Thiểu sản xương hàm dưới (còn gọi là tật cằm nhỏ )
- Tai ngoài thường nhỏ và có hình dạng bất thường , có thể xuất hiện các mẩu thịt thừa trước tai.
- Dị tật ống tai
- Dị tật xương con trong tai
- Dị tật các ống bán khuyên của tai trong
- Hở hàm ếch (có thể xảy ra trên một số bệnh nhân), góp phần làm tăng nguy cơ nhiễm trùng tai và gây mất thính lực
- Tịt cửa mũi sau gây ra các vấn đề về hô hấp
Các đặc điểm khác bao gồm bất thường tim, dị tật ngón tay cái và tầm vóc thấp. Một số bệnh nhân còn bị teo thực quản , trong đó đoạn trên của thực quản không nối liền với đoạn dưới và dạ dày. Phần lớn trẻ sơ sinh mắc teo thực quản (EA) cũng có rò khí quản-thực quản (TEF), triệu chứng gây sự kết nối bất thường giữa thực quản và khí quản. Sự kết nối này cho phép dịch từ thực quản đi vào đường thở và cản trở hô hấp. Teo thực quản/rò khí-thực quản (EA/TEF) nếu không được can thiệp điều trị sẽ cản trở quá trình ăn uống và gây tổn thương phổi do dịch thực quản liên tục đi vào đường thở.
Độ phổ biến
Loạn sản xương hàm mặt và tật đầu nhỏ là bệnh di truyền hiếm gặp với tỉ lệ mắc bệnh chưa được xác định. Hiện nay, hơn 60 trường hợp mắc bệnh đã được ghi nhận.
Nguyên nhân
Loạn sản xương hàm mặt và tật đầu nhỏ bắt nguồn từ các đột biến trên gen EFTUD2. Gen này cung cấp hướng dẫn để tạo ra một tiểu đơn vị của hai phức hợp gọi gồm thể cắt nối chính (major spliceosomes) và thể cắt nối phụ (minor spliceosomes). Thể cắt nối xử lí ARN thông tin (mRNA)—phân tử giữ vai trò như bản thiết kế di truyền để tạo ra protein. Thể cắt nối nhận biết rồi loại bỏ các vùng intron nhằm tạo ra phân tử mRNA trưởng thành.
Các đột biến gen EFTUD2 dẫn đến suy giảm số lượng hoặc mất chức năng của enzyme được tạo ra từ một bản sao của gen trong mỗi tế bào. Tình trạng thiếu enzyme này có khả năng làm suy yếu quá trình xử lí mRNA. Mối liên hệ giữa các đột biến này và những triệu chứng của bệnh vẫn chưa rõ ràng.
Chẩn đoán
Quá trình chẩn đoán bệnh thường trải qua nhiều bước và đòi hỏi sự tỉ mỉ trong đánh giá. Bác sĩ bắt đầu bằng quan sát và đánh giá các biểu hiện lâm sàng đặc trưng. Sau đó, xét nghiệm di truyền được thực hiện nhằm xác nhận kết quả chẩn đoán.
Trong quá trình chẩn đoán, một số bệnh lí có triệu chứng tương tự bao gồm:
- Hội chứng Treacher Collins
- Hội chứng Nager
- Hội chứng CHARGE
- Các rối loạn thuộc phổ dị tật mắt-tai-cột sống
Đối với các gia đình đã xác định được đột biến gen gây bệnh, họ có thể thực hiện xét nghiệm tiền sản trong những lần mang thai tiếp theo nhằm phát hiện sớm nguy cơ và có kế hoạch phù hợp.
Điều trị
Ngay sau sinh, một số trẻ mắc bệnh có thể cần can thiệp đường thở thông qua đặt nội khí quản hoặc mở khí quản. Các dị tật vùng sọ mặt đòi hỏi phương pháp điều trị riêng biệt cho từng trẻ, dưới sự phối hợp của nhiều chuyên khoa khác nhau.
Quá trình điều trị cần bao gồm các liệu pháp vận động, vật lí và ngôn ngữ để giúp trẻ phát triển tốt nhất. Bên cạnh đó, trẻ cũng cần được áp dụng các phương pháp điều trị tiêu chuẩn nếu xuất hiện các biến chứng như mất thính lực hay dị tật tim mạch.
Dạng di truyền
Loạn sản xương hàm mặt và tật đầu nhỏ di truyền trội trên nhiễm sắc thể thường, nghĩa là một bản sao của gen đột biến trong mỗi tế bào là đủ để gây ra bệnh. Phần lớn các trường hợp bắt nguồn từ đột biến mới (de novo) và xảy ra trên những người không có tiền sử gia đình mắc bệnh.
Trong các trường hợp khác, người bệnh thừa hưởng đột biến từ cha hoặc mẹ. Cha hoặc mẹ có thể mắc bệnh hoặc không ảnh hưởng. Đôi khi, cha hoặc mẹ chỉ mang đột biến gen trong một số hoặc tất cả các tế bào tinh trùng hoặc trứng, trường hợp được gọi là thể khảm dòng mầm. Trong những trường hợp này, cha mẹ không có dấu hiệu hoặc triệu chứng của bệnh.
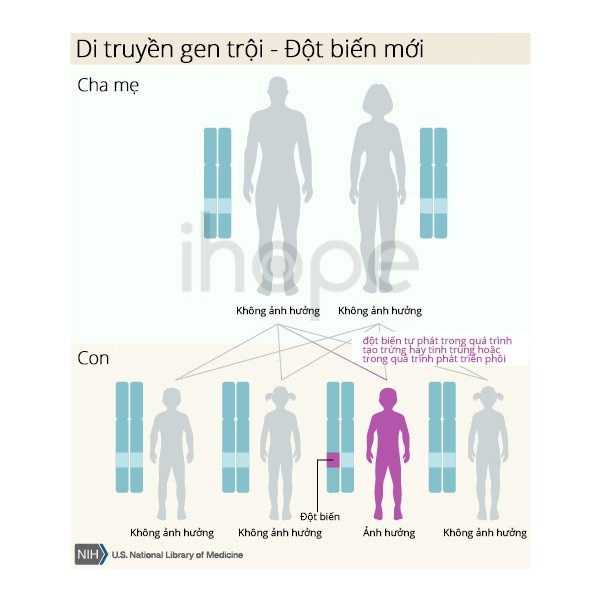
Nguồn: U.S. National Library of Medicine
Phòng ngừa
Loạn sản xương hàm mặt kèm tật đầu nhỏ di truyền trội trên nhiễm sắc thể thường, nếu cha hoặc mẹ mắc bệnh, con sinh ra sẽ có 50% khả năng di truyền bệnh. Do đó, để bảo đảm 100% khả năng con không bị bệnh, cha mẹ có thể chọn phương pháp thụ tinh nhân tạo IVF và sàng lọc phôi PGS/PGD. Các thành viên trong gia đình nên khám sức khỏe và tầm soát bệnh định kì nếu có thành viên mắc bệnh. Các cặp vợ chồng trước khi mang thai cần tư vấn và xét nghiệm di truyền đảm bảo sinh con khỏe mạnh.
Các tên gọi khác
- Mandibulofacial dysostosis, Guion-Almeida type
- MFDGA
- MFDM
References
- National Library of Medicine. Mandibulofacial dysostosis-microcephaly syndrome. Retrieved September 21, 2025 from https://www.ncbi.nlm.nih.gov/gtr/conditions/C1864652/
- Genetic and Rare Diseases Information Center. Central core myopathy. Retrieved September 21, 2025 from https://rarediseases.info.nih.gov/diseases/10056/index
- OMIM. MANDIBULOFACIAL DYSOSTOSIS, GUION-ALMEIDA TYPE; MFDGA. Retrieved September 21, 2025 from https://omim.org/entry/610536
- MedlinePlus. Mandibulofacial dysostosis with microcephaly. Retrieved September 21, 2025 from https://medlineplus.gov/genetics/condition/mandibulofacial-dysostosis-with-microcephaly/
- Orphanet. Mandibulofacial dysostosis with microcephaly. Retrieved September 21, 2025, from https://www.orpha.net/en/disease/detail/79113





