Loạn sản xương trán (frontometaphyseal dysplasia) là bệnh lý liên quan đến bất thường trong quá trình phát triển xương. Bệnh bao gồm các triệu chứng của hội chứng Otopalatodigital loại 1, hội chứng Otopalatodigital loại 2, hội chứng Melnick-Needles và loạn sản xương đầu cuối. Phần lớn người bệnh mất thính giác do dị tật xương con trong tai, các vấn đề phát triển vòm miệng và bất thường xương ngón tay, ngón chân.
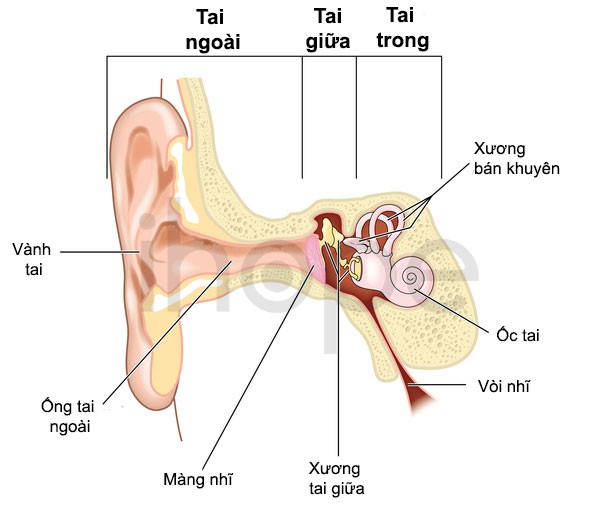
Cấu trúc tai
Nguồn: U.S. National Library of Medicine
Biểu hiện lâm sàng
Loạn sản xương trán được phân biệt với các bệnh lý khác bởi triệu chứng biến dạng khớp gây co rút hạn chế cử động. Người bệnh thường bị cong chi, vẹo cột sống và các ngón tay hoặc bàn tay bất thường.
Khuôn mặt của người bệnh có những đặc điểm bất thường như:
- Đôi mắt to và xếch xuống
- Gờ lông mày nổi rõ
- Mũi rộng, tẹt
- Hàm dưới và cằm nhỏ
- Thiếu răng hoặc răng mọc lệch
- Mất thính lực
Ngoài những bất thường về xương, người bệnh có thể bị tắc nghẽn niệu quản , dị tật tim, co thắt phế quản gây ra các vấn đề về hô hấp.
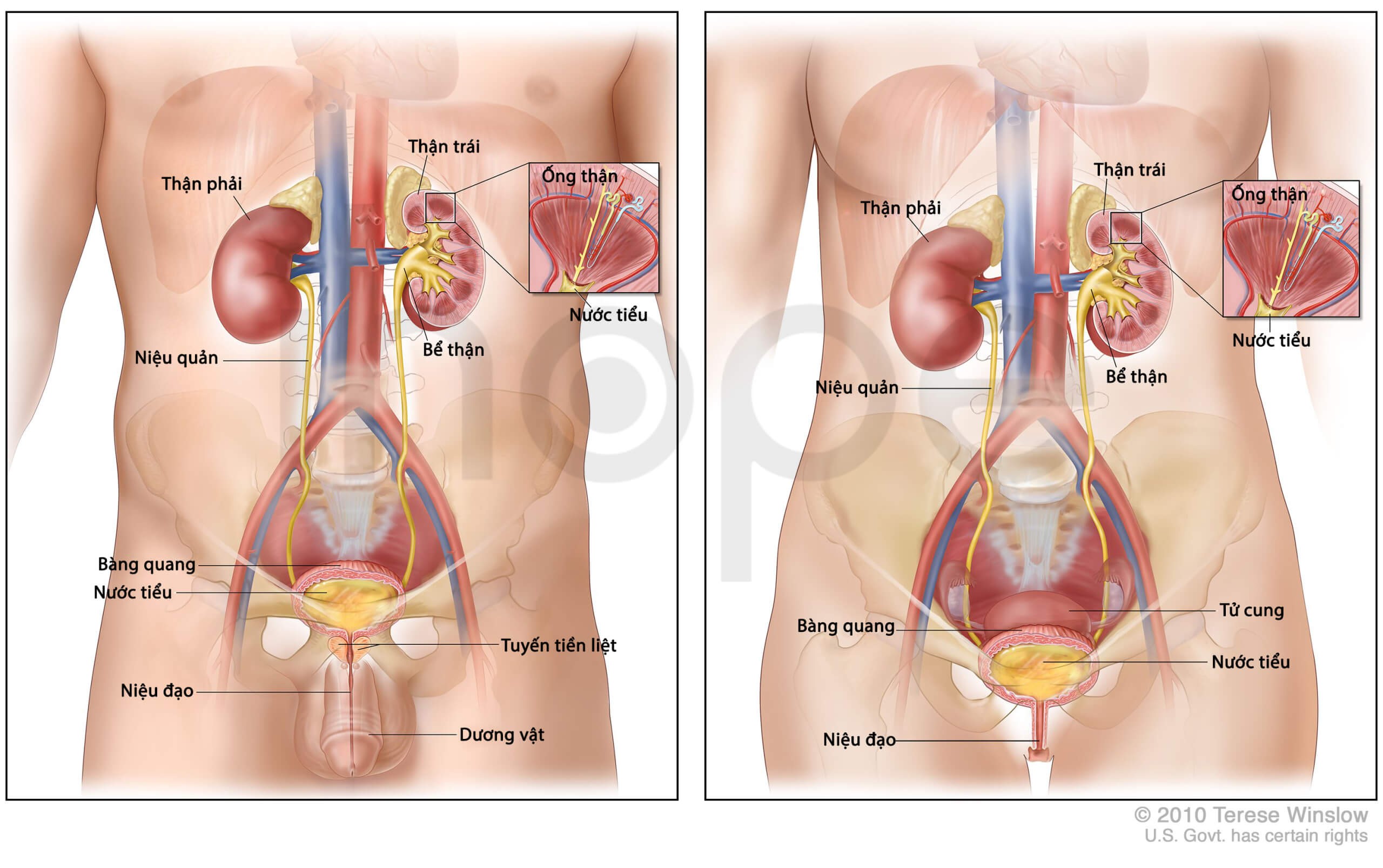
Ảnh: Giải phẫu thận nam và nữ
Nguồn: U.S National Library of Medicine
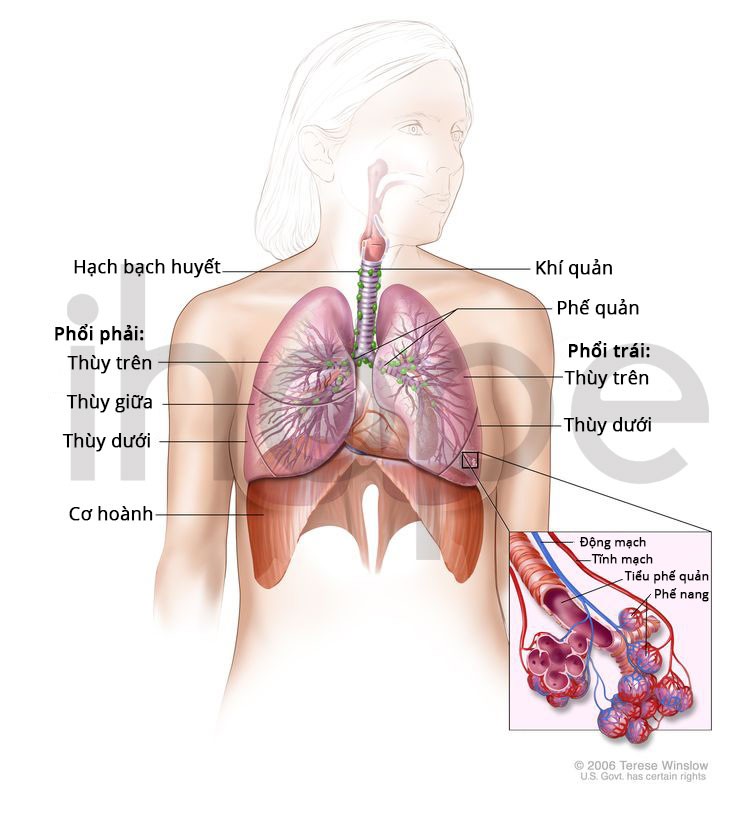
Ảnh: Cấu trúc hệ hô hấp
Nguồn: National Cancer Institute
Độ phổ biến
Loạn sản xương trán hiếm gặp, do đó hiện nay tỷ lệ mắc bệnh vẫn chưa được thống kê cụ thể.
Nguyên nhân
Loạn sản xương trán được chia thành 3 loại dựa vào nguyên nhân và dạng di truyền:
- Đột biến gen FLNA gây ra loại 1
- Đột biến gen MAP3K7 gây ra loại 2
- Đột biến gen TAB2 gây ra loại 3
Đột biến gen FLNA gây ra loạn sản xương trán. Gen FLNA cung cấp hướng dẫn sản xuất protein filamin A. Filamin A liên kết với actin để hình thành mạng lưới phân nhánh của các sợi tạo nên khung tế bào. Với cấu trúc này, tế bào có khả năng thay đổi hình dạng khi di chuyển.
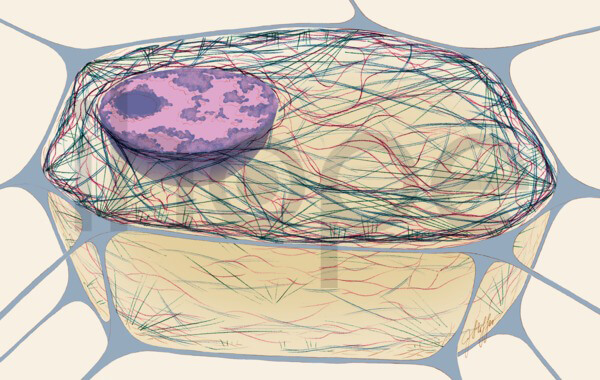
Ba sợi của khung tế bào bao gồm vi ống màu xanh lam, sợi trung gian màu đỏ và actin màu xanh lá cây
Nguồn: Judith Stoffer
Đột biến gen FLNA gây ra loạn sản xương trán làm thay đổi vùng liên kết của protein filamin A, dẫn đến tăng cường liên kết với actin. Các nhà nghiên cứu cho rằng các đột biến làm giảm tính ổn định của khung tế bào và phá vỡ quá trình phát triển xương, nhưng người ta chưa hiểu rõ vì sao những thay đổi trong protein gây ra triệu chứng của loạn sản xương trán.
Chẩn đoán
Chẩn đoán hội chứng loạn sản xương trán dựa trên biểu hiện lâm sàng và kết quả chụp X-quang. Xét nghiệm di truyền tìm đột biến gen FLNA giúp chẩn đoán bệnh.
Người mẹ mang đột biến gen FLNA có thể sàng lọc thai nhi bằng cách sinh thiết nhau thai hoặc chọc ối. Hai phương pháp này đều lấy ADN của thai nhi để chẩn đoán trước sinh nhưng thời điểm thực hiện khác nhau. Ngoài ra, siêu âm cũng có thể phát hiện các đặc điểm hình thái của thai nhi.
Điều trị
Hiện nay chưa có phương pháp điều trị loạn sản xương trán. Phần lớn trường hợp, bác sĩ điều trị dựa vào triệu chứng cụ thể của mỗi người bệnh. Trẻ mắc bệnh cần được hỗ trợ hô hấp ngay sau sinh. Vấn đề điều trị thính giác có thể hạn chế do vùng dị tật khó tiếp cận.
Dạng di truyền
Loạn sản xương trán xảy ra do biến thể trong gen FLNA di truyền theo kiểu trội liên kết với nhiễm sắc thể giới tính X. Một bản sao của gen bị đột biến trong mỗi tế bào đủ để gây ra bệnh. Một đặc điểm của di truyền liên kết nhiễm sắc thể giới tính X là người cha không thể truyền bệnh cho con trai. Nam giới mắc chứng loạn sản xương trán loại 1 thường có biểu hiện lâm sàng nghiêm trọng hơn nữ giới mắc bệnh.

Nguồn: U.S. National Library of Medicine
Đột biến trong gen MAP3K7 hoặc TAB2 gây ra loạn sản xương trán di truyền theo kiểu trội nhiễm sắc thể thường. Một bản sao của gen bị đột biến trong mỗi tế bào đủ để gây ra bệnh.

Nguồn: U.S. National Library of Medicine
Phòng ngừa
Bệnh di truyền liên kết X khó phát hiện ở những người phụ nữ mang gen bệnh cho đến khi sinh con. Để chủ động phòng ngừa, cha mẹ nên làm xét nghiệm sàng lọc gen lặn để chủ động cho tương lai của con. Các cặp vợ chồng trước khi mang thai cần tư vấn và xét nghiệm di truyền đảm bảo sinh con khỏe mạnh.
Trường hợp bệnh do đột biến gen MAP3K7 hoặc TAB2, khi sinh con sẽ có 50% khả năng di truyền bệnh. Do đó, để bảo đảm 100% khả năng con không bị bệnh, cha mẹ có thể chọn phương pháp thụ tinh nhân tạo IVF và sàng lọc phôi PGS/PGD.
Các tên gọi khác
- FMD
References
- Genetic Testing Information. Frontometaphyseal dysplasia 1. Retrieved January 17, 2023 from https://www.ncbi.nlm.nih.gov/gtr/conditions/C4281559/
- Genetic and Rare Diseases Information Center. Frontometaphyseal dysplasia. Retrieved January 17, 2023 from https://rarediseases.info.nih.gov/diseases/826/frontometaphyseal-dysplasia
- Catalog of Genes and Diseases from OMIM. FRONTOMETAPHYSEAL DYSPLASIA 1. Retrieved January 17, 2023 from https://omim.org/entry/305620
- U.S National Library of Medicine. Frontometaphyseal dysplasia. Retrieved January 17, 2023 from https://medlineplus.gov/genetics/condition/frontometaphyseal-dysplasia/
- MalaCards. Frontometaphyseal Dysplasia (FMD). Retrieved January 17, 2023 from https://www.malacards.org/card/frontometaphyseal_dysplasia
- National Organization for Rare Disorders. Frontometaphyseal dysplasia. Retrieved January 17, 2023 from https://rarediseases.org/gard-rare-disease/frontometaphyseal-dysplasia/
- Orphanet. Frontometaphyseal dysplasia. Retrieved January 17, 2023 from https://www.orpha.net/consor/cgi-bin/OC_Exp.php?lng=EN&Expert=1826
- Giuliano F, Collignon P, Paquis-Flucklinger V, Bardot J, Philip N. A new three-generational family with frontometaphyseal dysplasia, male-to-female transmission, and a previously reported FLNA mutation. Am J Med Genet A. 2005 Jan 15;132A(2):222. doi: 10.1002/ajmg.a.30396
- Morava E, Illes T, Weisenbach J, Karteszi J, Kosztolanyi G. Clinical and genetic heterogeneity in frontometaphyseal dysplasia: severe progressive scoliosis in two families. Am J Med Genet A. 2003 Jan 30;116A(3):272-7. doi: 10.1002/ajmg.a.10831
- Robertson SP. Otopalatodigital syndrome spectrum disorders: otopalatodigital syndrome types 1 and 2, frontometaphyseal dysplasia and Melnick-Needles syndrome. Eur J Hum Genet. 2007 Jan;15(1):3-9. doi: 10.1038/sj.ejhg.5201654