Loạn sản đa đầu xương (multiple epiphyseal dysplasia) là bệnh lí ảnh hưởng đến quá trình phát triển sụn, xương, chủ yếu là phần đầu xương dài tại tay và chân. Những bất thường này có thể gây ra nhiều triệu chứng liên quan đến khớp, từ đó người bệnh giảm khả năng vận động. Các biểu hiện thường khởi phát từ giai đoạn trẻ nhỏ và có thể tiến triển thành bệnh viêm khớp khởi phát sớm.
Biểu hiện lâm sàng
Phần lớn người bệnh được chẩn đoán từ giai đoạn trẻ nhỏ. Tuy nhiên, một số trường hợp mắc bệnh nhẹ chỉ được phát hiện khi đã trưởng thành. Loạn sản đa đầu xương được phân thành hai dạng dựa trên kiểu di truyền trội hoặc lặn.
Những triệu chứng chung của bệnh bao gồm:
- Đau khớp hông, đầu gối
- Viêm khớp khởi phát sớm
- Dáng đi lạch bạch
Mặc dù một số người mắc loạn sản đa đầu xương có tầm vóc thấp khi trưởng thành, phần lớn bệnh nhân vẫn có chiều cao bình thường.
Người mắc bệnh thể lặn biểu hiện thêm các triệu chứng khác như:
- Dị tật đầu gối
- Ngón tay, ngón chân cong bất thường
- Bàn chân khoèo
- Vẹo cột sống
- Hở hàm ếch
- Sưng tai
- Xương bánh chè chia thành hai mảnh
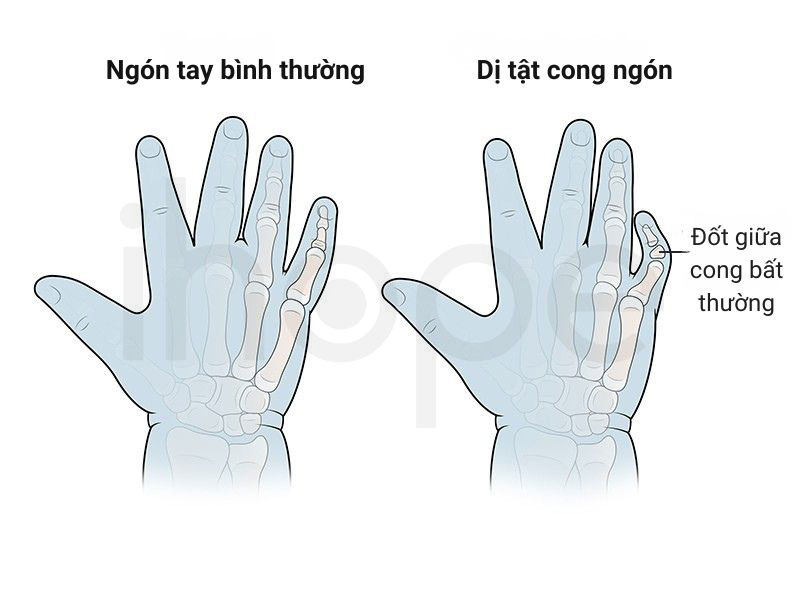
Ảnh: Dị tật cong ngón tay
Nguồn: Boston Children's Hospital
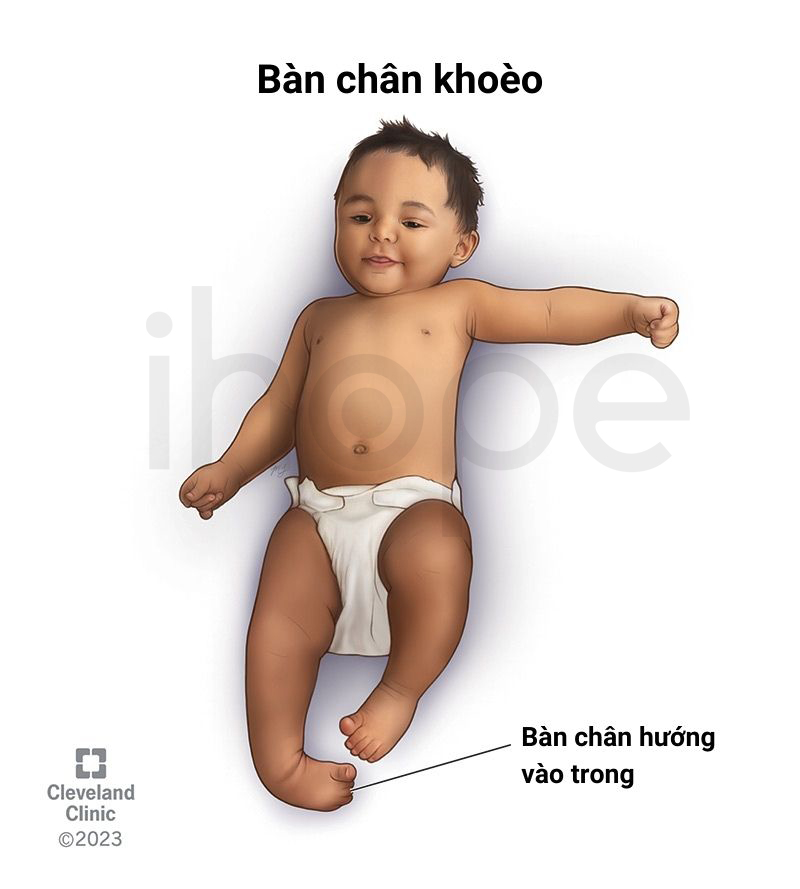
Ảnh: Bàn chân khoèo
Nguồn: Cleveland Clinic
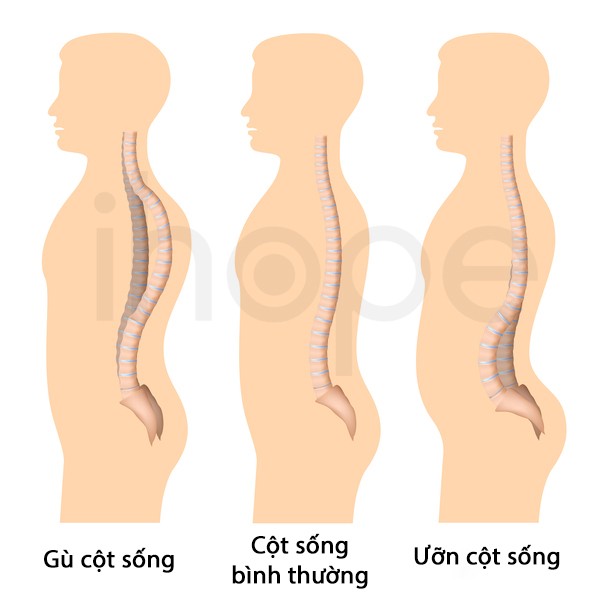 Ảnh: Cong cột sống
Ảnh: Cong cột sốngNguồn: Alila Medical Media

Ảnh: Hở hàm ếch
Nguồn: Health Centers for Disease Control and Prevention
Độ phổ biến
Ước tính tỉ lệ mắc loạn sản đa đầu xương thể trội trên toàn thế giới ít nhất khoảng 1/10.000 người. Tỉ lệ mắc loạn sản đa đầu xương thể lặn vẫn chưa được xác định rõ. Tuy nhiên, tỉ lệ mắc bệnh trong thực tế có thể cao hơn so với thống kê hiện tại do nhiều trường hợp có triệu chứng nhẹ không được chẩn đoán chính thức.
Nguyên nhân
Loạn sản đa đầu xương thể trội
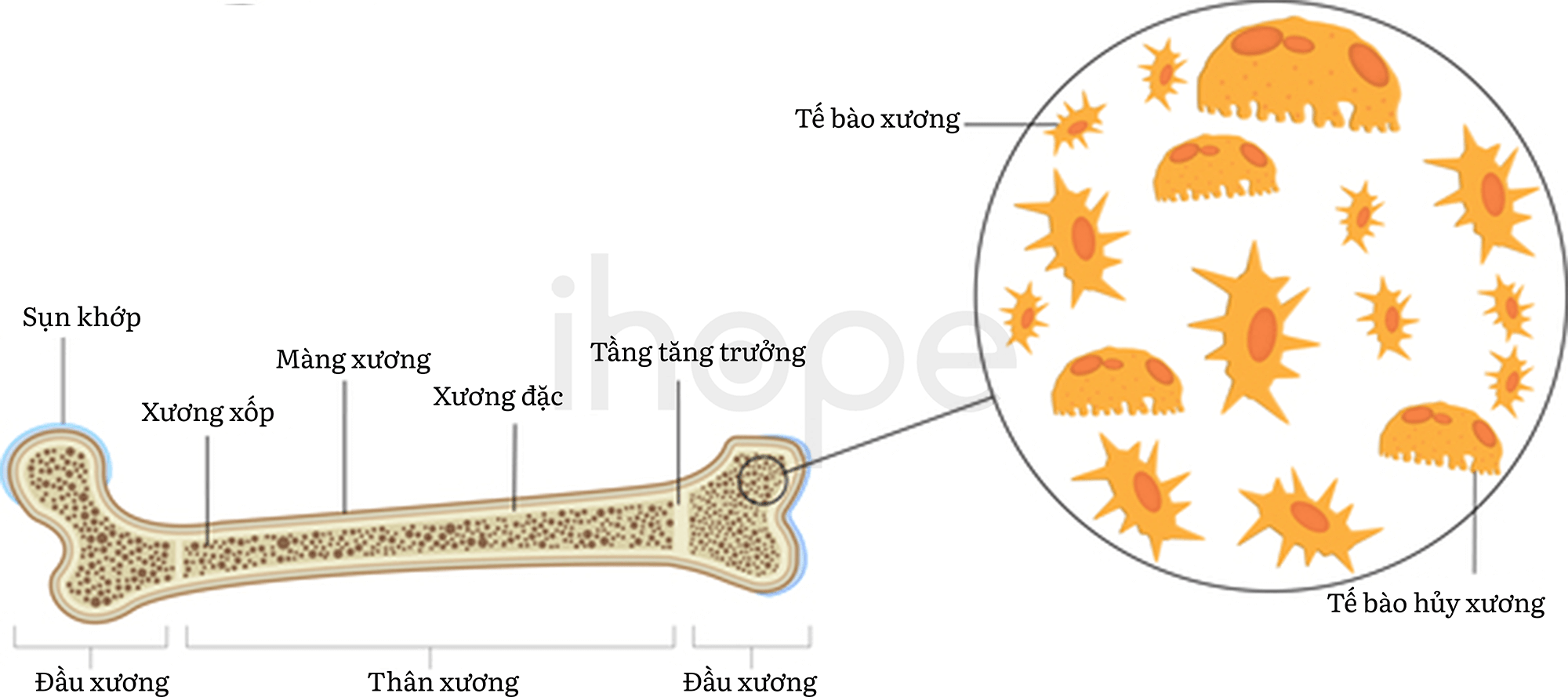
Loạn sản đa đầu xương thể trội có thể bắt nguồn từ đột biến trên nhiều gen khác nhau. Trong đó, gen COMP và MATN3 cung cấp hướng dẫn tạo ra protein giữ vai trò trọng yếu trong quá trình hình thành xương và sụn. Sụn là mô cứng, linh hoạt với chức năng tạo nên phần lớn bộ xương trong giai đoạn đầu phát triển phôi thai. Sau đó, các mô sụn sẽ chuyển thành xương, ngoại trừ phần đầu xương, mũi và tai ngoài. Đột biến gen COMP chiếm phần lớn các trường hợp thể trội và khoảng 10% trường hợp do đột biến gen MATN3 gây ra. Những đột biến này cản trở quá trình giải phóng protein vào khoảng gian bào giữa các tế bào tạo sụn, dẫn đến mô sụn hình thành bất thường, từ đó nhiều triệu chứng đặc trưng của bệnh loạn sản đa đầu xương thể trội khởi phát.
Nguồn: joshya/Shutterstock.com
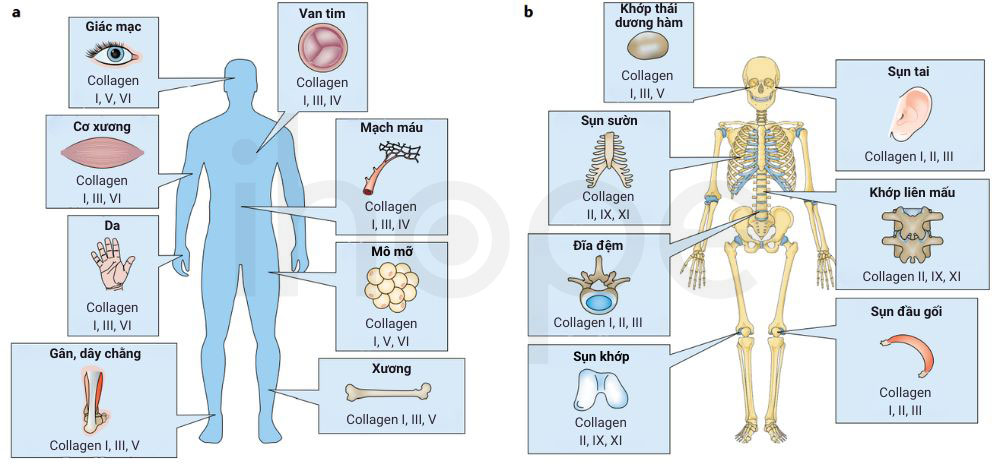
Ngoài ra, người ta ghi nhận bệnh thể trội có liên quan đến các gen COL9A1, COL9A2, COL9A3 cung cấp hướng dẫn tạo ra collagen loại IX—protein hỗ trợ tăng độ bền cho nhiều mô liên kết như da, xương, sụn, gân và dây chằng. Đột biến xảy ra trên các gen COL9A1, COL9A2 và COL9A3 chỉ được ghi nhận trong chưa đến 5% số người mắc loạn sản đa đầu xương thể trội. Hiện nay, người ta chưa rõ các đột biến này dẫn đến biểu hiện bệnh như thế nào. Tuy nhiên, một số nghiên cứu cho rằng chúng có thể khiến collagen loại IX tích tụ nhiều bên trong tế bào hoặc tương tác sai lệch với những thành phần khác của mô sụn.
Nguồn: Nature
Ngoài ra, trong một số trường hợp mắc loạn sản đa đầu xương thể trội, người ta không ghi nhận đột biến nào thuộc các gen trên cũng như chưa thể xác định rõ nguyên nhân gây bệnh.
Loạn sản đa đầu xương thể lặn
Đột biến gen SLC26A2 là nguyên nhân chính gây ra loạn sản đa đầu xương thể lặn. Gen này cung cấp hướng dẫn tạo ra một loại protein cần thiết cho quá trình phát triển bình thường của mô sụn và giai đoạn chuyển đổi sụn thành xương. Khi đột biến gen SLC26A2 xảy ra, cấu trúc sụn đang phát triển bị thay đổi, điều này cản trở quá trình hình thành xương đúng cách và dẫn đến các triệu chứng của bệnh loạn sản đa đầu xương thể lặn.
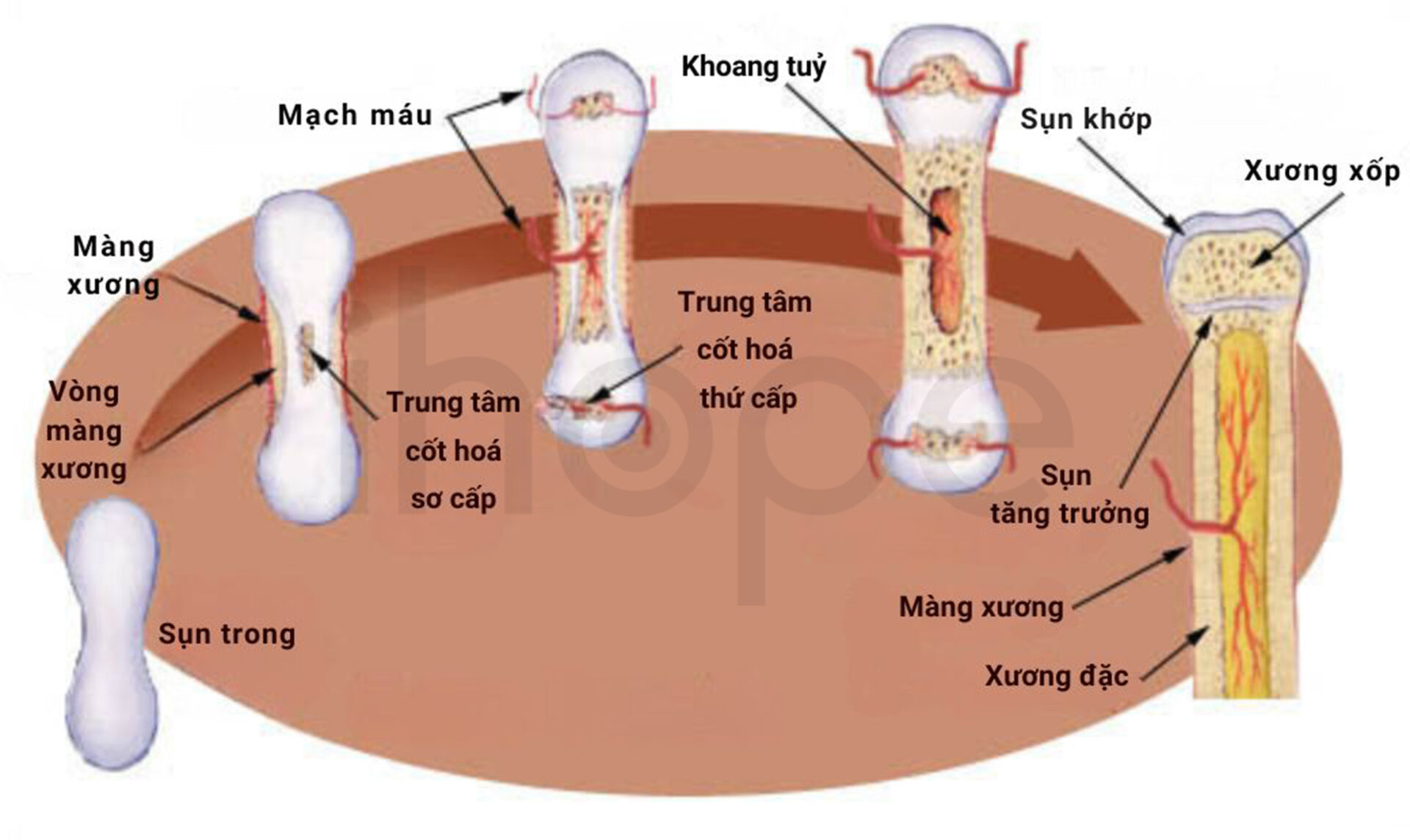
Nguồn: National Cancer Institute
Chẩn đoán
Loạn sản đa đầu xương được chẩn đoán dựa trên biểu hiện lâm sàng, kết quả chẩn đoán hình ảnh và xét nghiệm di truyền.
Chẩn đoán hình ảnh
Bệnh nhân được chỉ định chụp X-quang nhằm phát hiện các dấu hiệu sau:
- Chậm cốt hóa tại đầu xương
- Hình dạng và độ dài xương bất thường
- Tăng mật độ xương
- Dị dạng cột sống
- Thoái hóa khớp
Xét nghiệm di truyền
Bác sĩ thực hiện các xét nghiệm di truyền nhằm xác định những đột biến gen liên quan đến loạn sản đa đầu xương thể trội và lặn, qua đó kết quả chẩn đoán được xác nhận.
Điều trị
Hiện nay, chưa có phương pháp điều trị hoàn toàn loạn sản đa đầu xương. Các liệu pháp chủ yếu nhằm giảm nhẹ triệu chứng và cải thiện chất lượng đời sống người bệnh.
Một số biện pháp điều trị bao gồm:
- Sử dụng thuốc giảm đau
- Vật lí trị liệu
- Cắt xương, phẫu thuật đầu gối hoặc hông
Dạng di truyền
Loạn sản đa đầu xương di truyền theo kiểu trội trên nhiễm sắc thể thường. Do đó, một bản sao của gen đột biến trong mỗi tế bào đủ để gây ra bệnh. Người bệnh thường được di truyền từ cha hoặc mẹ mắc bệnh. Một số ít trường hợp là do đột biến mới (de novo) xuất hiện trong quá trình hình thành tế bào sinh sản (trứng, tinh trùng) hoặc quá trình phát triển phôi sớm.

Nguồn: U.S. National Library of Medicine
Loạn sản đa đầu xương cũng có thể di truyền theo kiểu lặn trên nhiễm sắc thể thường. Do đó, bệnh chỉ biểu hiện khi có cả hai bản sao của gen đột biến trong mỗi tế bào. Bệnh nhân mắc bệnh lặn trên nhiễm sắc thể thường

Nguồn: U.S. National Library of Medicine
Phòng ngừa
Loạn sản đa đầu xương di truyền trội trên nhiễm sắc thể thường, nếu cha hoặc mẹ mắc bệnh, con sinh ra sẽ có 50% khả năng di truyền bệnh. Trường hợp di truyền lặn do đột biến gen lặn, cha mẹ mang đột biến dị hợp nên gần như không có biểu hiện bệnh, do đó rất khó phát hiện cho đến khi sinh con. Do đó, để bảo đảm 100% khả năng con không bị bệnh, cha mẹ có thể chọn phương pháp thụ tinh nhân tạo IVF và sàng lọc phôi PGS/PGD. Các thành viên trong gia đình nên khám sức khỏe và tầm soát bệnh định kỳ nếu có thành viên mắc bệnh. Các cặp vợ chồng trước khi mang thai cần tư vấn và xét nghiệm di truyền nhằm đảm bảo sinh con khỏe mạnh.
Các tên gọi khác
- EDM1
- EDM2
- EDM3
- EDM4
- EDM5
- Epiphyseal dysplasia, Fairbank type
- Epiphyseal dysplasia, multiple, 1
- Epiphyseal dysplasia, multiple, 2
- Epiphyseal dysplasia, multiple, 3
- Epiphyseal dysplasia, multiple, 4
- Epiphyseal dysplasia, multiple, 5
- Epiphyseal dysplasia, Ribbing type
- MED
- Multiple epiphyseal dysplasia, autosomal dominant
- Multiple epiphyseal dysplasia, autosomal recessive
- RMED
References
- U.S National Library of Medicine. Multiple epiphyseal dysplasia. Retrieved August 6, 2025 from https://medlineplus.gov/genetics/condition/multiple-epiphyseal-dysplasia/
- National Organization for Rare Disorders. Autosomal Dominant Multiple Epiphyseal Dysplasia. Retrieved August 6, 2025 from https://rarediseases.org/rare-diseases/dominant-multiple-epiphyseal-dysplasia/
- National Organization for Rare Disorders. Recessive Multiple Epiphyseal Dysplasia. Retrieved August 6, 2025 from https://rarediseases.org/rare-diseases/recessive-multiple-epiphyseal-dysplasia/
- U.S National Library of Medicine. Multiple Epiphyseal Dysplasia, Autosomal Dominant. Retrieved August 6, 2025 from https://www.ncbi.nlm.nih.gov/books/NBK1123/
- Orphanet. Multiple epiphyseal dysplasia. Retrieved August 6, 2025 from https://www.orpha.net/en/disease/detail/251